


Полная версия
Фармакология может быть доступной. Иллюстрированное пособие для врачей и тех, кто хочет ими стать



На кажущийся объем распределения оказывает влияние связывание с белками плазмы и депонирование. Так, липофильные вещества (например, неингаляционное средство для наркоза тиопентал) перераспределяются из плазмы в жировую ткань и, накапливаясь там, значительно увеличивают значение Vd.
Биодоступность (f) – это показатель, описывающий поступление ЛС в системный кровоток при используемом пути введения по сравнению с внутривенным введением. Это часть введенного лекарственного вещества, достигшая системного кровотока относительно введенной дозы, выраженная в процентах. Препараты, вводимые внутривенно, имеют биодоступность 100 %.
Используя временной показатель, можно рассчитать площадь под кривой (AUC) на графике зависимости концентрации от времени. Отношение (AUC перорально /AUC в\в) x 100 позволяет определить биодоступность (f).
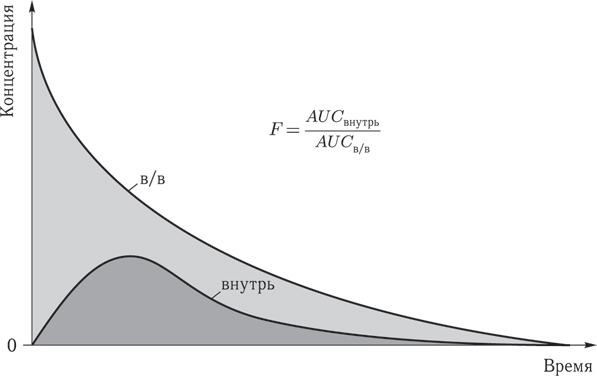
Рис. 2. Биодоступность (f) – это часть введенного лекарственного вещества, выраженная в процентах, достигшая системного кровотока относительно введенной дозы. (Пояснения в тексте.)
Метаболизм ЛС происходит подобно другим ксенобиотикам, попавшим в организм. ЛС метаболизируются во многих тканях, но преимущественно в печени, и этот процесс подразумевает превращение молекулы лекарства в легко выводимые метаболиты. Основная цель метаболизма лекарств – облегчение выведения чужеродных химических соединений за счет повышения их растворимости в воде (гидрофильности). Очевидно, что ферменты, метаболизирующие лекарства, развились как универсальная защита от чужеродных химических веществ, поступающих из окружающей среды.
Гидрофильные вещества легче выводятся с мочой, так как не подвергаются реабсорбции в почечных канальцах. При этом химические модификации в процессе метаболизма значительно чаще снижают, чем увеличивают фармакологическую активность ЛС. Метаболически активируемые, но исходно неактивные ЛС называются пролекарствами (например, активный метаболит антиагреганта клопидогрел образуется под воздействием фермента CYP2С19, входящего в систему микросомальных ферментов печени).
Некоторые ЛС в значительной степени или полностью инактивируются при первом прохождении через печень, в связи с чем их необходимо вводить сублингвально или парентерально. Так, биодоступность нитроглицерина при приеме внутрь составляет менее 1 % из-за эффекта первого прохождения через печень. Однако при сублингвальном приеме, при котором нитроглицерин всасывается, минуя воротную вену и печень, этот показатель возрастает до 38 %.
Основными эффекторами метаболизма лекарств являются ферменты цитохрома Р450 (CYP450) в печени.
Выделяют две основные фазы метаболизма. Большинство реакций фазы I осуществляют всего несколько монооксигеназ широкого спектра действия подсемейства CYP (цитохром Р450) 1–3, так называемых микросомальных ферментов печени. Наиболее часто в метаболизме ЛС участвует фермент CYP3A4. Фаза I метаболизма состоит из реакций восстановления, окисления или гидролиза. Реакции фазы I обычно превращают исходное лекарство в более полярный метаболит посредством образования групп – ОН, – NH2 или – SH.
Недостаточно полярные для элиминации препараты могут быть впоследствии (или первично) модифицированы ферментами фазы II, характеризующейся синтетическими реакциями. Наиболее частыми реакциями фазы II являются конъюгации с глюкуроновой кислотой. Кроме того, ЛС могут быть конъюгированы с глутатионом или глицином или модифицированы путем переноса метильных, ацетильных или сульфагрупп от соединений-доноров.
Выделяют два основных типа кинетики метаболизма лекарств. Кинетика нулевого порядка: скорость метаболизма и/или элиминации остается постоянной и не зависит от концентрации препарата в плазме в равновесном состоянии (Cплазма снижается линейно с течением времени).
Нулевой порядок – это метаболизм с ограниченной емкостью [этанол, фенитоин, аспирин (в высоких концентрациях)].
Кинетика первого порядка: скорость метаболизма и/или элиминации прямо пропорциональна концентрации препарата в плазме (Cплазма экспоненциально снижается с течением времени).
Первый порядок – это метаболизм, зависящий дозы (большинство ЛС; рис. 3).
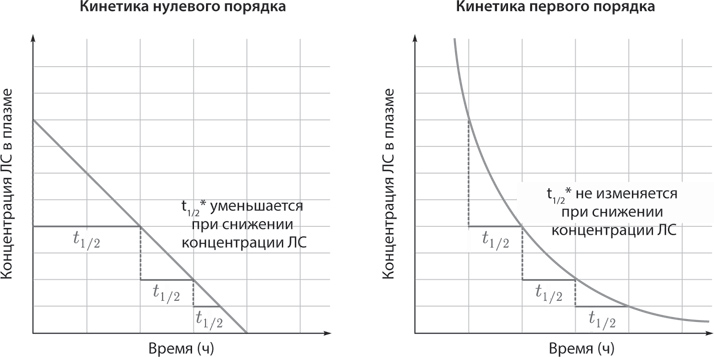
Рис. 3. Типы кинетики метаболизма лекарственных средств. (Пояснения в тексте.) (* – период полувыведения, см. стр. 31)
Активность микросомальных ферментов печени может изменяться. Важной причиной снижения индивидуальной активности микросомальных ферментных систем являются инактивирующие мутации (полиморфизмы) генов, кодирующих микросомальные ферменты печени. Так, существуют аллели CYP2C19, кодирующие образование фермента со сниженной или отсутствующей функцией (обладатели таких полиморфизмов являются так называемыми слабыми метаболизаторами). У таких пациентов со сниженной активностью CYP2C19 при приеме пролекарства клопидогрела меньшее количество препарата переходит в активную форму, что проявляется его сниженной эффективностью. В результате возникает риск тромбоза коронарных сосудов на фоне формально правильно назначенных ЛС. Активность ферментов может снижаться на фоне препаратов, ингибирующих их активность (например, макролиды и коназолы), а также у пациентов с заболеваниями печени или недостаточным печеночным кровотоком. Метаболизм ЛС ускоряется у пациентов, получавших индукторы ферментов печени, метаболизирующих лекарственные препараты (фенобарбитал, зверобой продырявленный). Индукция и ингибирование метаболизма лекарств являются примерами фармакокинетических взаимодействий лекарств.
Скорость и интенсивность метаболизма ЛС варьирует не только среди пациентов, но даже у одного конкретного человека с течением времени (с этим связаны ограничения в дозах ЛП у детей и пациентов старше 60 лет).
Выведение, осуществляемое преимущественно почками, подразумевает удаление ЛС и его метаболитов из организма.
Почки являются основным (но не единственным) органом, выделяющим лекарство. Три компонента почечной экскреции: клубочковая фильтрация, секреция и реабсорбция – определяют скорость этого процесса. Лекарственные вещества, экскретируемые почками, попадают в мочу посредством клубочковой фильтрации. Кроме того, некоторые лекарственные средства выделяются в проксимальные канальцы переносчиками органических катионов и анионов. Липофильные препараты дополнительно секретируются в проксимальные канальцы посредством пассивной диффузии, так как они легко проникают через мембраны клеток нефрона.
Пассивная диффузия является основным механизмом реабсорбции лекарств из нефрона. В наименьшей степени реабсорбция характерна для гидрофильных, т. е. полярных или ионизированных препаратов, из-за их неспособности проникать через мембраны клеток нефрона. Уменьшение реабсорбции за счет увеличения гидрофильности является основной задачей печеночного метаболизма лекарств. Кроме того, физиологические или индуцированные изменения значения pH мочи изменяют реабсорбцию лекарственных средств, способных к ионизации. Например, случайная передозировка ацетилсалициловой кислоты (аспирина) лечится инфузией бикарбоната. Возникающее в результате ощелачивание мочи благоприятствует образованию ионизированной формы аспирина, которая не способна к реабсорбции путем пассивной диффузии, поэтому увеличивается скорость выведения ЛС.
У большинства людей почечная экскреция ЛС с возрастом снижается главным образом из-за физиологического снижения клубочковой фильтрации, а также в результате различных заболеваний почек. Во избежание токсичности, зависящей от накопления, у таких пациентов необходимо соответственно уменьшить дозу препаратов, выводимых с мочой и характеризующихся низким терапевтическим индексом. Усиление выведения ЛС, в свою очередь, наблюдается при уменьшении связывания с белками плазмы, поскольку только несвязанная фракция лекарственного средства подвергается фильтрации и секреции. Типичная ситуация – совместное введение двух препаратов, конкурирующих за одни и те же сайты связывания с белками плазмы (см. раздел «Распределение препаратов» выше). При нормальной функции почек избыток несвязанного ЛС быстро выводится из организма без накопления и токсичности.
Выделение ЛС и метаболитов с желчью (и, соответственно, с калом), выдыхаемым воздухом, слюной и грудным молоком играет значительно меньшую роль по сравнению с почечной эксрецией. Исключение составляет выведение летучих средств для наркоза с выдыхаемым воздухом. Особое внимание уделяется экскреции с грудным молоком, так как оно может накапливать алкалоиды и липофильные соединения и оказывать токсическое воздействие на младенца, находящегося на грудном вскармливании.
Клиренс ЛС (CL) является показателем скорости элиминации препарата. Это показатель объема плазмы, который очищается от препарата за единицу времени. Общий клиренс большинства лекарств представляет собой сумму клиренса, выводимого почками, и метаболизма в печени и других органах.
CL = Vd x Ke, где
Vd = объем распределения,
Ke = константа элиминации (Ke =0,693\t1/2),
CL = скорость выведения/концентрация в плазме.
Период полувыведения (t½) – это время, необходимое для того, чтобы концентрация препарата в плазме достигла половины своего исходного значения.
В клинической практике важным параметром является равновесная концентрация (steady state), т. е. динамическое равновесие между поступлением и выведением ЛС. При этом концентрация лекарства остается постоянной, потому что скорость выведения лекарства равна скорости введения лекарства.
При кинетике ЛС первого порядка
t½ = (0,693 х Vd) / CL,
где Vd – кажущийся объем распределения,
CL – клиренс ЛС.
Предположим, что мы вводим дозу ЛС каждый период полувыведения. Тогда
• После дозы 1 в конце интервала дозирования осталось 0,5 дозы. Это означает, что мы находимся в устойчивом состоянии на 50 %.
• После дозы 2 в организме находится 1,5 дозы, затем половина выводится, остается 0,75 дозы (75 % равновесного состояния).
• После дозы 3 в организме находится 1,75 дозы, затем половина выводится, остается 0,875 дозы (88 % равновесного состояния).
• После дозы 4 в организме находится 1,875 дозы, затем половина выводится, остается 0,9375 дозы (94 % равновесного состояния).
• После дозы 5 в организме находится 1,9375 дозы, затем половина выводится, остается 0,96875 (97 % устойчивого состояния).
Считается, что при 97 % система находится в приблизительно устойчивом состоянии, где скорость поступления равна скорости выведения одной дозы за интервал дозирования.
Дозы лекарственных веществ
Различают терапевтические, токсические и летальные дозы.
Терапевтические дозы подразделяются на
• минимальные терапевтические (действующие);
• средние терапевтические;
• высшие терапевтические.
Минимальные терапевтические (действующие) дозы (пороговые дозы) вызывают минимальный терапевтический эффект. Обычно они в 2–3 раза меньше средней терапевтической дозы.
Средние терапевтические дозы оказывают на большинство больных необходимое фармакотерапевтическое действие.
Ударная доза – доза, превышающая среднюю терапевтическую дозу.
С нее обычно начинают лечение противомикробными средствами (антибиотиками, сульфаниламидами) для быстрого создания высокой концентрации вещества в крови. После достижения определенного терапевтического эффекта назначают поддерживающие дозы.
При длительном применении ЛВ указывается его доза на курс лечения (курсовая доза).
Высшие терапевтические дозы – предельные дозы, превышение которых может привести к развитию токсических эффектов. Их назначают, если применение средних доз не оказывает желаемого действия.
Токсические дозы – дозы, оказывающие токсическое действие на организм.
Летальные дозы (от лат. letum – смерть) – дозы, вызывающие смертельный исход (определяется на стадии доклинических испытаний).
Диапазон доз от минимальной действующей до минимальной токсической определяется как широта терапевтического действия. Чем она больше, тем безопаснее применение ЛС.
Повторное введение лекарственных веществ
Повторные введения одного и того же ЛВ могут приводить к количественному (увеличение или уменьшение) и качественному изменению фармакологических эффектов.
Явления, возникающие при повторных введениях ЛС
Кумуляция (от лат. cumulatio – увеличение, скопление) – накопление в организме ЛВ или вызываемых им эффектов.
Материальная кумуляция – увеличение в крови и/или тканях содержания ЛВ после каждого последующего введения по сравнению с предыдущим. При повторных введениях могут накапливаться ЛВ, медленно инактивируемые и выводимые из организма, а также ЛВ, прочно связывающиеся с белками плазмы крови или депонирующиеся в тканях, например некоторые снотворные средства из группы барбитуратов, препараты наперстянки. Материальная кумуляция может быть причиной токсических эффектов, что необходимо учитывать при дозировании таких препаратов.
Функциональная кумуляция – усиление эффекта ЛВ при повторных введениях при отсутствии повышения его концентрации в крови и/или тканях. Этот вид кумуляции может возникать при длительных повторных приемах алкоголя. При развитии алкогольного психоза («белая горячка») у восприимчивых лиц бред и галлюцинации развиваются в то время, когда этиловый спирт уже метаболизировался и не определяется в организме.
Сенсибилизация. Многие ЛВ образуют комплексы с белками плазмы крови, которые приобретают при определенных условиях антигенные свойства. Это сопровождается образованием антител и сенсибилизацией организма. Повторное введение тех же ЛВ вызывает аллергические реакции. Часто такие реакции возникают при повторных введениях пенициллинов, прокаина, водорастворимых витаминов, сульфаниламидов.
Привыкание (толерантность, от лат. tolerantia – терпение) – уменьшение фармакологического эффекта ЛВ при его повторных введениях в той же дозе. При развитии привыкания для достижения прежнего эффекта необходимо увеличивать дозу ЛВ.
Привыкание к ЛВ, стимулирующим рецепторы (к агонистам рецепторов), может быть обусловлено снижением чувствительности (десенситизацией) рецепторов и/или уменьшением их плотности (количества).
Привыкание к некоторым ЛВ связано с изменением их фармакокинетики (уменьшением всасывания, увеличением скорости метаболизма и/или выведения). Так, основной причиной привыкания к фенобарбиталу считают активацию его метаболизма вследствие индукции ферментов печени, вызываемой самим фенобарбиталом.
Частный случай привыкания – тахифилаксия (от греч. tachys – быстрый, phylaxis – защита) – быстрое развитие привыкания при повторных введениях препарата через короткие промежутки времени (10–15 мин.).
Лекарственная зависимость. Это настоятельная потребность (непреодолимое стремление) в постоянном или периодически возобновляемом приеме определенного ЛВ или группы веществ. Вначале вещество принимают для достижения состояния эйфории, благополучия и комфорта, устранения тягостных переживаний, испытания новых ощущений. Однако через определенное время потребность в повторном приеме становится непреодолимой, что усугубляется синдромом отмены: возникновением при прекращении приема данного вещества тяжелого состояния, связанного с психическими и соматическими нарушениями (нарушениями функций органов и систем организма). Такое состояние обозначают термином «абстиненция» (от лат. abstinentia – воздержание).
Различают психическую и физическую лекарственную зависимость.
Психическая лекарственная зависимость характеризуется резким ухудшением настроения и эмоциональным дискомфортом, ощущением усталости при лишении препарата. Она возникает при применении кокаина и других психостимуляторов (амфетамина), галлюциногенов (диэтиламид лизергиновой кислоты, LSD-25), никотина, индийской конопли (анаша, гашиш, план, марихуана).
Физическая лекарственная зависимость характеризуется не только эмоциональным дискомфортом, но и возникновением синдрома абстиненции – состояния, включающего объективные и субъективные нарушения.
Физическая лекарственная зависимость развивается к опиоидам (героину, морфину), барбитуратам, бензодиазепинам, алкоголю (этиловому спирту).
Вопрос
Правильно ли я понял, что привыкание и лекарственная зависимость совсем не одно и то же?
Да, это так. Привыкание, снижение эффективности ЛС, с течением времени характерно при приеме многих групп лекарственных препаратов и может развиваться как на уровне синаптической передачи (эфедрин), так и на системном уровне (антигипертензивные средства). Лекарственная зависимость, непреодолимое желание к повторному приему ЛС характерна для небольшого числа ЛС, которые подлежат особому учету и должны назначаться в течение ограниченного времени. Возможно, смешение этих понятий связано с тем, что для опиоидов, вызывающих тяжелую лекарственную зависимость, характерно развитие зависимости и привыкания.
Фармакодинамика
Фармакодинамика – это изучение механизмов действия, посредством которого лекарства производят свои фармакологические эффекты.
Важнейшим разделом фармакодинамики является рецепторная теория, которая прошла долгий и сложный путь развития. Пауль Эрлих выдвинул постулат в отношении концепции рецепторов: corpora non agunt nisi ixata (вещества не действуют, пока не свяжутся), – и первым сравнил взаимодействие вещества и рецептора с ключом и замком. Уверенность в универсальности своей формулы остановила П. Эрлиха в шаге от открытия гематоэнцефалического барьера. Он ввел краситель метиленовый синий внутривенно мышам и обнаружил, что все органы, кроме мозга, окрасились в синий цвет. Он сделал вывод, что в мозге нет рецепторов к метиленовому синему. Лишь позже его ученик Э. Голдман ввел краситель в мозг мыши и увидел, что тот окрасился в синий цвет, а остальные органы – нет.
Основными субстратами для лекарственных средств в организме являются рецепторы, ферменты, транспортные системы и каналы.
Рецепторы, взаимодействующие с лекарственными средствами, представляют собой специализированные макромолекулы-мишени (в основном это белковые молекулы – липопротеины, гликопротеины, нуклеопротеины и др.), которые связываются с эндогенными медиаторами, гормонами, аутокоидами. Лекарственные средства, имеющие сродство (аффинитет) с этими рецепторами, действуют либо подобно эндогенным биологически активным веществам, либо блокируют их действие.
Рецептор выполняет двойственную роль: он должен распознать связывающуюся с ним молекулу и преобразовать полученный сигнал в ответ. В соответствии с этими задачами рецептор имеет распознающий домен и эффекторный домен. Образование комплекса ЛС – рецептор приводит к биологической реакции. Величина ответа пропорциональна количеству комплексов ЛС – рецептор.
Молекулы (например, ЛС, гормоны, медиаторы), которые связываются с рецептором, называются лигандами. Способность лиганда связываться с данным рецептором зависит от его сродства (аффинности) к рецептору, а способность активировать рецепторы и приводить к клеточному ответу зависит от внутренней активности лиганда. Сродство и активность лекарства определяются его химической структурой и не зависят друг от друга. Таким образом, степень, в которой лиганд способен вызывать возбуждение рецептора, ведущее к клеточному ответу, называется внутренней активностью.
Вопрос
Почему рецептор и лиганд связываются друг с другом?
Рецептор является белковой молекулой, состоящей из различных аминокислот, имеющих неоднородное электрическое поле. Это приводит к образованию структуры, имеющей уникальное расположение электрических зарядов. Соответственно, лиганд будет иметь противоположные заряды, позволяющие связаться с участком рецептора. В случае обратимой связи рецептор-лиганд взаимодействие обеспечивается ионными, водородными, гидрофобными, Ван-дер-Ваальсовыми силами. При необратимом связывании между рецептором и лигандом возникает ковалентная связь.
Лиганды в зависимости от результата связывания с рецептором делятся на агонисты и антагонисты.
Агонисты активируют рецепторы, вызывая желаемый ответ. Агонист, который вызывает максимальный ответ, называется полным агонистом, его внутренняя активность принимается за 1. Однако не все агонисты способны вызывать максимальный ответ. Если активность лигандов, например ЛС, меньше 1, такие ЛС называют частичными агонистами. Многие гормоны, медиаторы (например, ацетилхолин, гистамин, норадреналин) и ЛС (например, пилокарпин, ксилометазолин, бензодиазепины, окситоцин) действуют как агонисты.
Антагонисты – это группа ЛС, которые предотвращают активацию рецепторов. Предотвращение активации рецепторов может вызывать много фармакологических эффектов. Так, антагонисты усиливают клеточную функцию, если они блокируют действие какой-либо тормозной системы организма, которое обычно снижает клеточную функцию (так, блуждающий нерв угнетает активность синоатриального узла сердца и уменьшает частоту сердечных сокращений; атропин блокирует рецепторы, через которые осуществляется угнетение, и за счет этого увеличивает частоту сердечных сокращений). В то же время антагонисты снижают клеточную функцию, если они блокируют действие вещества, которое обычно повышает клеточную функцию (М-холиноблокатор атропин снижает стимулирующее действие ацетилхолина на секрецию слюнных желез, что приводит к снижению секреции и ощущению «сухости во рту»).
Выделяют несколько типов антагонистов.
Конкурентный антагонизмЕсли агонист и антагонист конкурируют за связывание с одним и тем же рецептором, то антагонист называют конкурентным (блокатор). При этом взаимодействие агониста и антагониста дозозависимо: увеличение дозы агониста позволяет преодолеть блокаду, вызванную антагонистом, и получить прежний эффект при условии большей концентрации.
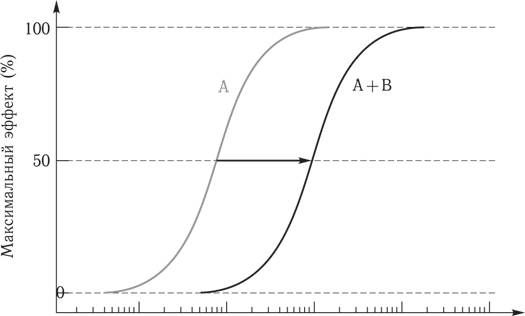
Рис. 4. Образование комплекса лекарство – рецептор приводит к биологической реакции. Величина ответа пропорциональна количеству комплексов лекарство – рецептор. Обычный способ представления взаимосвязи между концентрацией лекарственного средства и биологической реакцией – это кривая «доза (концентрация) – эффект». Пример конкурентного антагонизма. Агонист А вызывает определенный эффект. Если он вводится на фоне антагониста (В), то прежний 100 % эффект достигается в большей концентрации
Неконкурентный антагонизм
При этом виде антагонизма ЛС-антагонист связывается с участком рецептора, отличным от сайта связывания агониста, но изменяет структуру сайта связывания агониста и таким образом снижает сродство агониста. Практически при введении неконкурентного антагониста уменьшается число рецепторов, с которыми может связаться агонист. В результате эффективность агониста снижается, и увеличение концентрации агониста ее уже не восстановит.
Вопрос
Непонятно, куда исчезают рецепторы при неконкурентном антагонизме? Здесь написано, что их становится меньше.
Все рецепторы остаются на месте, но неконкурентный антагонист связывается с рецептором Х, а изменяет сродство рецептора Y к агонисту, который перестает с ним, рецептором Y, связываться. Поэтому даже если увеличить концентрацию агониста, эффект не возрастет, так как связаться с рецептором Y нет возможности.
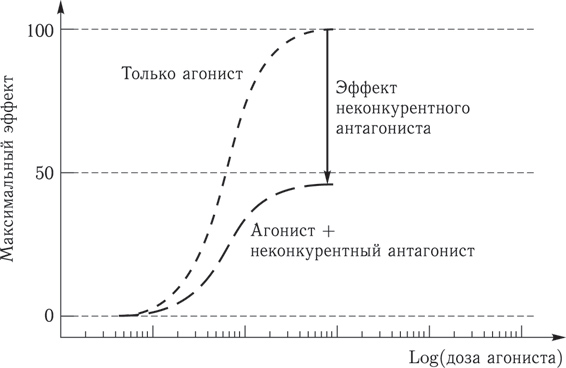
Рис. 5. Неконкурентный антагонизм. Неконкурентный антагонист связывается с иным рецептором, чем рецептор агониста. При этом уменьшается число рецепторов, с которыми может связаться агонист
Функциональный (физиологический) антагонизм
При этом типе антагонизма две различные молекулы действуют на разные рецепторы и производят физиологически противоположные эффекты.


