


Полная версия
Хроническая алкогольная интоксикация


Действие метаболитов этилового алкоголя
Выделенный метаболический аспект токсического действия этилового алкоголя связан с его биотрансформацией, продукты которой являются «четвертой силой», разрушающей клетки изнутри.
Этанол блокирует гексокиназную ферментную систему, которая обеспечивает проникновение глюкозы в клетки и переход ее в глюкозо-6-фосфат. Глюкозу потребляют клетки всех тканей, поэтому при ее недостатке возникает гипогликемия и создаются предпосылки для снижения интенсивности гликолиза и цикла Кребса. Головной мозг и эритроциты особенно чувствительны к гипогликемии, поэтому в организме человека существуют дополнительные источники получения глюкозы на случай чрезвычайной ситуации. Такими источниками являются депо гликогена в печени и ресинтез глюкозы из ее предшественников: аминокислот, лактата и глицерина, которые имеются в каждой группе клеток. Подчеркнем, что все же главным источником поступления глюкозы в организм является пища.
Метаболические последствия в виде гипогликемии, возникающей даже после умеренного однократного приема этилового спирта, частично компенсируются перечисленными биохимическими механизмами. Острая интоксикация этанолом вызывает «биохимическую бурю», а частое употребление спиртных напитков можно сравнить с постоянно действующей чрезвычайной ситуацией для промежуточного обмена веществ.
Во-первых, принятая с пищей глюкоза не усваивается из-за блокады гексокиназы, «да и есть не особенно хочется», поскольку 1 г этанола поставляет около 7 Ккал (для сравнения: это калораж, затрачиваемый на 150 м пути, если грести по воде сидя в лодке). Однако «алкогольные» калории не аккумулируются в макроэрги-ческих связях и дают только тепло.
Во-вторых, при биотрансформации этилового алкоголя в рассматриваемых условиях дополнительные источники получения глюкозы либо блокируются (как, например, глюконеогенез), либо истощаются (как, например, гликогенолиз). В-третьих, при активации перечисленных метаболических путей гормонами, на фоне постоянного присутствия этанола в плазме крови даже в незначительных количествах, образуются агрессивные субстраты, которые депонируются в печени, миокарде, головном мозге и нарушают их функции.
Представленная цепь событий отражает суть возникающих нарушений только в общих чертах. Более глубокий анализ патохимических нарушений при хронической алкоголизации позволяет выделить их ключевые звенья, на которые могут быть направлены усилия фармакотерапии.
Первым метаболитом этанола является ацетоальдегид, который превосходит по токсичности исходную молекулу. Ацетоальдегид преимущественно образуется в гепатоцитах при участии алкоголь-дегидрогеназ (см. рис. 4).
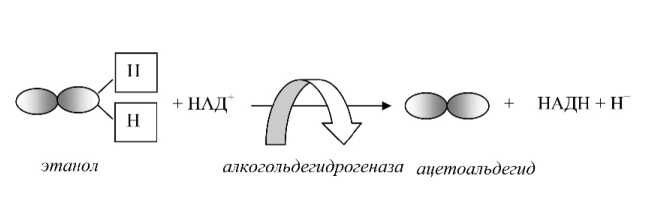
Рис. 4. Образование ацетоальдегида и дефицита НАД+
Условные обозначения:
НАД+ – никотинамиддинуклетотидфосфат;
НАДН – никотинамиддинуклетотидфосфат (восстановленная форма).
Второй протон при восстановлении НАД+ высвобождается в среду. Дефицит НАД+ и одновременное накопление ионов водорода создают условия для развития ацидоза.
Нужно сказать, что все дегидрогеназы переносят электроны и протоны, образующиеся в окислительно-восстановительных реакциях, поэтому они нуждаются в ко-ферментах, т. е. в промежуточных переносчиках функциональных групп. Наиболее важным из них является НАД+-амидникотиновой кислоты, который передает восстановительный эквивалент в дыхательную цепь и участвует в аккумуляции энергии в макроэргах. В результате образования ацетоальдегида НАД+ восстанавливается в НАДН + Н+3, таким образом в ходе реакции биодеградации этилового спирта НАД+ потребляется.
Далее из ацетоальдегида образуется уксусная кислота. Реакция ее образования также протекает с участием альдегиддегидрогеназ, что означает повторное потребление НАД+.
Следовательно, в результате двух метаболических реакций, которые претерпевает этанол в клетках печени, дважды снижается концентрация НАД+.
Дефицит НАД+ сопровождается: блокадой глюконеогенеза; замедлением гликолиза и цикла Кребса; остановкой бета-окисления и отложением жира в печени; изменением поляризации клеточных мембран различных органов за счет сдвига соотношения НАДН/ НАД+.
В результате этого углубляется гипогликемия и возникает дефицит энергии. Клиническими эквивалентами этих изменений являются: слабость, разбитость, «плохое самочувствие» в целом, чувство тяжести в области сердца, головная боль, озноб, гипергидроз, нарушение работы желудочно-кишечного тракта.
С целью компенсации уровня глюкозы плазмы крови распадается гликоген печени. Однако его запасы даже у здоровых людей невелики. Всего лишь на несколько часов гликогенолиз покрывает расход глюкозы нейронами мозга и эритроцитами крови (самыми энергоемкими потребителями), после чего его депо истощаются.
У голодных людей, у лиц, находящихся на диете, у больных диабетом, принимающих гипогликемические средства (за исключением толбутамида), у пациентов, принимающих гепатотоксичные препараты (парацетамол, изониазид, альдомет, фенотиазины и т. д.), бета-адреноблокаторы или средства для снижения массы тела, а также при повторном приеме алкоголя, – запасы гликогена в печени снижены, поэтому гипогликемия у них развивается стремительно, является тяжелой и длительной (Ионин М. Л., 1998). У алкоголиков запасы гликогена в печени снижены почти до нуля.
У здоровых, сытых и трезвых людей существует мощный механизм защиты от гипогликемии. Это синтез глюкозы de novo в печени и в почках. Он осуществляется из глюкогенных аминокислот, лактата и глицерина. Этот процесс называется глюконеогенезом, и его мощность составляет до 250 г глюкозы в сутки. Подчеркнем, что глюконеогенез является самым главным источником питания нейронов и эритроцитов, которые полностью зависят от постоянного интенсивного снабжения глюкозой. Однако для осуществления глюконеогенеза необходим НАД+, запасы которого у лиц, систематически употребляющих спиртное, истрачены на биотрансформацию этилового алкоголя. В этих условиях НАД+ отчуждается от других, порой «недублируемых», биохимических реакций (например, из реакций электронотранспортной цепи флавопротеинов), и они «останавливаются». Это усугубляет дефицит энергии и создает основу для гипоксии различных тканей.
Гликолиз должен осуществляться любой ценой, иначе клетки погибнут. Однако продолжающийся прием спиртного нарушает декарбоксилирование кетокислот и замыкает гликолиз на выходе. Тогда источником образования энергии становятся жиры (триацилглицериды). Они распадаются на жирные кислоты и глицерин. У человека жирные кислоты не могут трансформироваться в глюкозу, они поступают в кровь в неэстерифицированном состоянии (НЭЖК). Жирные кислоты – биохимически агрессивные субстраты – являются источником образования свободных радикалов, стимулируют высвобождение биологически активных веществ, поэтому существует путь их быстрой утилизации в печени, где они превращаются в ацетилкоэнзим-А. Концентрация последнего в печени резко увеличивается, его избыток конденсируется в ацето-уксусную кислоту, из которой образуются 3-гидроксибутират и ацетон, т. е. кетоновые тела. Возникают предпосылки для развития алкогольного кетоацидоза, опасного, рецидивирующего состояния, несвоевременное лечение которого может приводить к летальному исходу.
В свою очередь глицерин также не может трансформироваться в глюкозу (как это происходит у нормальных людей), поскольку этот процесс также катализируется дегидрогеназой, для работы которой также нужен НАД+. В этих условиях в печени вновь синтезируются нейтральные жиры, которые депонируются в гепатоцитах и создают предпосылки для развития их жировой дистрофии.
Таким образом, в условиях гипогликемии, спровоцированной этанолом, «сгорания жиров в пламени углеводов» не происходит, из них образуются кетоновые тела и депозиты жира в печени, ее границы увеличиваются, а функции угасают. Синтетические процессы, происходящие в ней в нормальном состоянии, изменяются, снижается антитоксическая функция печени, что приводит к появлению в крови большого количества аутокоидов, способных вызывать различные нарушения в организме больного.
В попытке повысить уровень глюкозы в плазме крови увеличивается содержание гормонов, регулирующих ее концентрацию, таких как кортизол, глюкагон, гормон роста, адреналин, и снижается секреция инсулина. Гормональные сдвиги практически не влияют на уровень глюкозы, так как ее неоткуда взять. Однако они усиливают липолиз, стимулируют дальнейшее отложение жира в печени и предоставляют материал для дальнейшего образования кетонов.
Сдвиг равновесия НАДН+Н+/НАД+ влево изменяет поляризацию мембран клеток. Потенциал клеточных мембран становится более негативным по сравнению с исходным потенциалом, что еще больше нарушает транспорт ионов через них. Страдают функции транспортных насосов, в том числе тех из них, которые призваны эвакуировать отработанные продукты обмена веществ. Снижение интенсивности обменного транспорта в системе К+/Н+ (антипорт «калий – водород») сопровождается накоплением избытка ионов водорода в клетках. К кетонемическому ацидозу присоединяется внутриклеточный метаболический ацидоз.
В совокупности все это приводит к срыву функции органов, который проявляется в соответствии с их физиологической ролью (нарушения ритма сердца, декомпенсация функций печени, органические дисфункции головного мозга ит. д.).
Однако представленная патохимическая драма не исчерпывается явлениями, связанными с дефицитом НАД+. Сами метаболиты этанола – ацетоальдегид и уксусная кислота, образующиеся при его биотрансформации, – завершают ее финал. Несмотря на то, что ацетоальдегид вырабатывается эндогенным путем во многих биохимических реакциях, превышение его концентрации в головном мозге оказывает выраженное нейротропное и гепато-токсическое действия (Зиматкин С. М., 1993). Ацетоальдегид необратимо связывает белки синаптосом в трофотропных системах ЦНС (например, в ГАМКергических), влияет на продукцию эндогенных опиатов, угнетает транспортные АТФазы мембран нейронов головного мозга (Багров А. Я., 1996), наконец, ацетоальдегид угнетает синтез ацетилхолина – главного функционального антагониста катехоламинергических (возбуждающих) систем головного мозга, индуцирует цитохром СУР 2Е1.
Физиологические пределы колебаний концентрации ацетоальдегида очень малы, его избыток должен немедленно утилизироваться в биохимических реакциях. Однако некоторые продукты биотрансформации ацетальдегида также оказывают повреждающее действие на ткани: например, при деградации ацетальдегида ксантиноксидазой или альдегидоксидазой образуются свободные радикалы. Связывание ацетальдегида с цистеином или глютатионом вызывает увеличение их оборота и потерю пула серосодержащих аминокислот (что также сопровождается накоплением пероксидов и повреждением митохондрий). Длительная алкоголизация и накопление ацетальдегида вызывают снижение пула эндогенного метионина и его активной формы S-аденозилметионина, который участвует в синтезе глютатиона (S-аденозилметионин является наиболее важным агентом в реакциях трансметилирования, которые обеспечивают пластический обмен веществ).
Вместе с тем ацетоальдегид способен усиливать активность ионотропных глицинергических рецепторов (Mascia, 2001). Продукты конденсации ацетоальдегида с норадреналином, серотонином, эндорфинами, такие, как сальсолинол, гармалин и другие, способны конкурентно взаимодействовать с рецепторами указанных лигандов и изменять характер нейро– и котрансмиссии в головном мозге. Возникает хаос в рецепторконтролируемых ионных каналах мембран нейронов.
Это обстоятельство влияет на многие системные процессы, включая функции ВНД, такие как мышление, эмоционально-волевые и другие реакции.
В последние годы установлено, что ацетоальдегид является мощным стимулятором продукции «медиаторов воспаления» интерлейкиновой (IL) группы цитоксинов: IL-6 (фактора некроза опухолей альфа), IL-8 и IL-1, локальные и системные эффекты которых связывают с прямым гепатоцитотоксическим действием, угнетением факторов роста, нейро– и гемопоэза (см. действие на органы), и с развитием демиелинизирующих процессов в головном мозге (Rothwell, 1995).
Уксусная кислота также агрессивна, и поэтому существует механизм ее быстрой утилизации при переходе в ацетилкоэнзим-А, который происходит в митохондриях печени. Реакция его образования высокоэнергетична и сопряжена с экзоэргическими процессами (после приема спиртного всегда становится жарко).
При хроническом алкоголизме метаболический переход ацетальдегида в ацетат снижается. Избыток ацетальдегида в полной мере проявляет представленные выше виды действия.
Еще раз подчеркнем, что избыток ацетилкоэнзима-А блокирует бета-окисление жирных кислот, а также усиливает синтез холестерина и тормозит скорость парциальных реакций цикла Кребса. Это способствует еще большему депонированию жира в печени.
Наряду с дегидрогеназами в биотрансформации этанола принимают участие каталаза и стимулированная этанолом микросомальная оксидаза (система МЭОС с участием цитохромов Р-450). Эти пути биотрансформации этанола «включаются» по достижении его уровня в плазме крови, – в среднем от 1‰ (100 мг%), и они служат способом дополнительной элиминации этого яда из ор га низ ма.
Негативные последствия биотрансформации этанола в системе МЭОС включают:
дополнительное образование ацетоальдегида;
деградацию ксенобиотиков и лекарственных препаратов;
продукцию свободных радикалов и усиление ПОЛ;
избыточный синтез коллагена;
образование аутоантител к продуктам деградации этанола.
При сформированной зависимости от этанола система МЭОС работает наравне с дегидрогеназной системой, увеличивая образование продуктов биотрансформации этилового спирта, т. е. биохимические нарушения в организме усугубляются.
Недегидрогеназная (неокислительная) биодеградация этанола приводит к образованию алкогольных эфиров жирных кислот – биологически активных метаболитов, которые появляются в плазме крови и во внутренних органах при длительной алкоголизации субъекта. Эти вещества оказывают токсическое действие на клетки органов, в которых они образуются.
Чем дольше длится алкоголизация, тем более глубокими являются представленные патохимические нарушения: нарастает функциональная недостаточность различных органов, т. е. возникает полиорганная недостаточность.
Поражения различных систем организма, возникающие при систематическом употреблении этилового алкоголя
Первичный контакт яда с организмом человека происходит на уровне ЖКТ, однако то действие, которое алкоголь оказывает на человека, к нему привычного, принципиально отличается от того действия, которое испытывают люди непьющие или мало употребляющие спиртные напитки. Это умозаключение было впервые высказано J. Pringsheim (1908), который показал, что у алкоголиков спиртные напитки всасываются и разрушаются быстрее, чем это происходит у непьющих людей. Это была одна из первых попыток, предложенных для объяснения явления толерантности к алкоголю, и в те годы было трудно предположить, какие глубокие нарушения гомеостаза у пьющего человека скрываются под вершиной этого «айсберга».
Все изменения, возникающие в органах при хронической интоксикации этанолом, взаимосвязаны друг с другом и происходят из-за действия этилового алкоголя на мембраны, рецепторы и обмен веществ, протекающий в клетках. Между тем в каждой физиологической системе имеются особенности этих изменений, обусловленные выполняемой ею функцией.
Действие на ЖКТ
Здесь следует выделить два фактора, которые влияют на токсикодинамику этанола при хронической интоксикации. В желудке начинается пресистемный метаболизм этанола (его разрушение алкогольдегидрогеназой, которая вырабатывается микробной флорой желудка) и всасывание этанола и ацетоальдегида.
При гастрите и язвенной болезни активность алкогольдегидрогеназы слизистой оболочки желудка увеличена, поэтому у таких лиц прием спиртного сопровождается усилением образования ацетальдегида. Могут возникать осложнения, связанные с его действием (лактацидемия, ацетатемия, выраженные вегетативные реакции, тяжелые алкогольтетурамовые реакции, возможно, и более быстрое развитие привыкания).
Заболевания желудка (болезнь Мэллори – Вейса, синдром Рэндю – Ослера, состояния после его резекции) снижают активность алкогольдегидрогеназы и интенсивность пресистемного метаболизма этанола. В результате эффект опьянения происходит от меньших количеств спиртного.
Препараты, угнетающие активность алкогольдегидрогеназы желудка (аспирин, парацетамол, Н-2-гистаминоблокаторы), замедляют биотрансформацию этанола и снижают токсическое действие, обусловленное ацетальдегидом, однако способствуют усилению опьянения.
Средние показатели активности алкогольдегидрогеназы у женщин вдвое ниже по сравнению с мужчинами. Поэтому при потреблении одинаковых количеств спиртного в плазме крови женщин концентрация этанола нарастает быстрее (иными словами, женщины пьянеют быстрее мужчин).
Частый прямой контакт этанола со слизистыми оболочками желудка, тощей и тонкой кишки вызывает в них дистрофические процессы: отек клеточных элементов, эксфолиацию, нарушение микроциркуляции, атрофию слизистых оболочек и образование язв. Эрозивный гастрит часто является причиной кровотечений из верхних отделов ЖКТ, которые возникают у 20 % хронических алкоголиков (Lawrence et al., 1996). Наиболее уязвимы дистальная часть 12-перстной кишки и проксимальная часть тощей кишки, поскольку в этих отделах равновесные концентрации этанола (в разделах кровь/ткань) удерживаются более длительно, по сравнению с другими отделами кишечника. Морфологические изменения слизистой оболочки сопровождаются нарушением пристеночного пищеварения, всасывания глюкозы, тиамина, фолиевой кислоты, цианкобаламина и, особенно, метионина, что является долнительным фактором в нарушении обмена веществ и развитии осложнений алкоголизма.
В указанных отделах кишечника локализованы гипофиз-независимые нейроэндокринные клетки, секретирующие в кровь вещества, аналоги которых в головном мозге выполняют медиаторные и другие функции. При иммунохимической оценке секреторной активности биоптатов дистального отдела нисходящей части 12-перстной кишки у здоровых непьющих добровольцев и пациентов с низким уровнем потребления спиртного (менее 40 г/неделю) была выявлена большая плотность рецепторов холецистокинина, галанина, ингибиторного пептида желудка, глюкагона, мотилина, нейропептида Y, питуитарного аденилатциклазо-стимулирующего пептида, секретина, серотонина, соматостатина, субстанции Р, ВИП-гормона, чем в биоптатах пьющих людей (Hauge et al., 2001).
Исследование механизмов химической зависимости по составляющим APUD-систему аутокоидам является весьма перспективным в силу доступности и возможности визуального, морфологического и гистохимического исследования этих отделов ЖКТ. Используя в качестве инструмента теорию тождества регуляторных систем «на входе и выходе» (в частности, холинореактивных систем (Денисенко П. П., 1982)) применительно к аутокоидам APUD-системы и рассматривая абстинентные синдромы, возникающие при отмене героина, клофелина, этанола, бета-адрено-блокаторов и т. д., в виде типовых патологических процессов, в рабочей гипотезе «о наркомании – болезни кишечника?» мы высказали предположение о возможной маркерной роли аутокоидов APUD-системы в установлении предрасположенности к действию наркотических веществ (Афанасьев В. В., с соавт., 1986).
Следует подчеркнуть, что всасывание этанола в желудке контролируется холинореактивными системами через М-холинорецепторы, причем участвуют в этом процессе все их подтипы, кроме М-1-рецепторов (Kinoshita, 2001).
При алкоголизации эрозируется слизистая оболочка желудка и тонкого кишечника. В совокупности с изменениями свертывающих свойств крови это обстоятельство может приводить к желудочно-кишечным кровотечениям.
Алкоголь также проявляет панкреатотоксическое действие. Обострение хронического панкреатита особенно часто возникает при приеме спиртного на фоне (или совместно) с лекарственными средствами, оказывающими панкреатотоксическое действие и при употреблении сахара («закусить конфеткой»).
Действие на ЦНС
Изменения в ЦНС являются следствием патохимических процессов, индуцированных систематическим приемом этилового алкоголя. Биохимические нарушения промежуточного, белкового и других видов обмена веществ, мембранотоксические эффекты, дисрегуляция рецепторного аппарата клеток и их ферментных систем, образование большого количества биологически агрессивных веществ в клетках ЦНС и их матриксе – играют роль в нейроадаптации нейронов головного мозга, нейрофармакологической сущностью которой является стойкое преобладание активности возбуждающих структур в ЦНС над тормозными, вплоть до глубокого угнетения функциональной активности послед них.
Исследованиями Э. А. Костандова с соавторами (1981) было установлено, что экспозиция этанолом сопровождается большим угнетением корковой активности правого полушария головного мозга. Авторы обратили внимание на сходство психопатологической продукции, возникающей при очаговых органических поражениях правого полушария головного мозга и хронической алкоголизации, таких как беззаботность, беспечность, благодушие, своеобразный юмор («алкогольный» юмор), нарушение критики и осознания своего заболевания и его последствий и другие признаки.
У лиц, хронически употребляющих этанол, практически во всех отделах мозга в той или иной степени имеются структурно-функциональные изменения: во фронтальной коре большого мозга, медиальных и темпоральных долях, гиппокампе, диэнцефалоне, переднем мозге. В меньшей степени задействованы стрио-паллидарные структуры. В перечисленных отделах мозга снижается региональный кровоток, возникают атрофические процессы, происходит редукция астроцитной глии и олигодендроцитов. Существенно снижается общий вес мозговой ткани (по данным 8735 аутопсий вес мозга алкоголиков в среднем на 37 г меньше веса мозга у лиц без алкогольной патологии (Torvik et al., 1982)). Нейро-патологические изменения мозга при аутопсии алкоголиков-муж-чин в возрасте от 30 до 64 лет выявлены в 67,7 % случаев (представлены результаты 194 аутопсий). Наиболее частыми из них были: энцефалопатия Вернике (14,2 %), церебральная атрофия (37 %), демиелинизация волокон Варолиевого моста (2 %), печеночная энцефалопатия (1 %), причем в 28 % случаев регистрировались сочетанные поражения (Skullerud et al., 1991).
Нарушаются когнитивные функции мозга, страдает память, концентрация внимания, изменяется эмоционально-волевая деятельность и тонкие социальные связи. Возникают внешние и внутренние конфликты, формируются варианты подкрепления, постепенно структурируются мотивация и элементы аддиктивного поведения. Между девиантным поведением детей, рожденных от родителей-алкоголиков, и электровозбудимостью нейронов фронтальной коры их головного мозга существует взаимосвязь, прослеживающаяся в поколениях. Корреляция между предрасположенностью таких подростков к алкоголизму и снижением амплитуды (и скрытого периода) поздних волн, вызванной электрической активностью головного мозга (так называемая волна Р300), у них настолько высока, что позволяет выделять изменение Р300 в качестве маркера этого заболевания.
Длительное и частое употребление этанола провоцирует развитие цереброваскулярной патологии. В ближайшее время после приема большого количества спиртного в среднем на сутки усиливается тромбообразование, которое может приводить к острому нарушению мозгового кровообращения. Спустя несколько суток после приема большого количества спиртного увеличивается частота геморрагических инсультов (Taylor, 1982), причем у женщин субарахноидальное кровоизлияние встречается чаще, чем у мужчин.
В основе геморрагических нарушений лежит волнообразное изменение синтеза биологических веществ в системе арахидоновой кислоты, возникающее при чередовании абстиненции и интоксикации, что приводит к волнообразному изменению реологических свойств крови. Другой причиной является либерализация этанолом определенной группы цитокинов в клетках оболочек головного мозга и его нейронах. В интервале 24–48 ч цитокины (особенно IL– 1b– и g-интерферон) увеличивают продукцию NO в этих клетках, что вызывает вазодилатацию (Shih, 2001).
В последние годы большое значение в развитии геморрагических инсультов при «алкогольной» болезни отводят гипомагнигистии и гипомагниемии. Раннее выраженное и длительное снижение концентрации этого иона в телах нейронов, особенно при сочетанной патологии (например, ААС и ЧМТ) сопровождается вазоспазмом церебральных сосудов, снижением креатинфосфата в клетках ЦНС, падением рН и редукцией фосфорилирующего потенциала цитозоля нейронов (отношение креатинфосфата к неорганическому фосфору). Эти клинические наблюдения были подтверждены интересным экспериментом Altura (1999) на животных, с использованием имплантатов мини-помпы для перфузии желудочков мозга раствором этанола, выполненных по типу ставших уже историческими опытов Feldberg (1956). Перфузия мозга 30 %-ным раствором этанола вызывала 30 %-ное снижение концентрации внутриклеточного магния, которое удерживалось вплоть до 14-го дня после окончания перфузии и сопровождалось падением уровня креатинфосфата и фосфорилирующего потенциала цитозоля на 15 и 40 %, соответственно, причем впоследствии у животных повышалась чувствительность к токсическому действию «малых» доз алкоголя, введение которых вызывало у них геморрагический инсульт.







