


Полная версия
How to Grow a Human
The fruit-fly embryo acquires its initial pattern features from morphogen threshold concentrations. Pretty much the first thing it does is to define which end will become the head and thorax, and which end the abdomen. In other words, the embryo acquires a front–rear axis. That is defined by a morphogen protein called bicoid. At the tip of the “head” (so-called anterior) end, the embryo produces bicoid, and this begins to diffuse down to the rear (posterior) end. The concentration falls smoothly from the anterior to the posterior end. Where it exceeds certain values, the bicoid protein will bind to the DNA within the embryo and activate other genes with vivid names like hunchback, sloppypaired 1 and giant (typically named because of the developmental defects that mutations in the genes can produce). How this switching occurs is complicated, not least because it also seems to depend on a gradient of another protein called caudal that diffuses from the opposite (posterior) end. But the outcome is that the embryo becomes quite sharply segmented into regions where different genes are expressed or not. Thus the uniformity of the embryo is destroyed: an anterior– posterior axis is established, along with the segments that will develop into the fly’s head, thorax and abdomen. It seems that similar gradients cause segmentation of the neural tube of vertebrates: the tissues that will become our brain and spinal column.
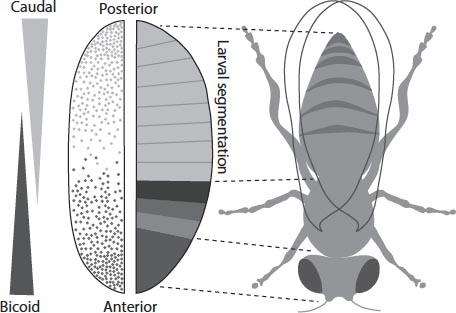
Gradients in the concentration of proteins bicoid and caudal from opposite ends of the fruit-fly embryo switch on genes at different positions that cause segmentation of the body plan.
Other diffusing morphogens produce other kinds of gradient, defining different axes of the emerging body. For example, a protein called dorsal is involved in setting up the top-to-bottom (dorsoventral) axis of the fruit-fly embryo that distinguishes the region that will become the back (where the larva will ultimately grow wings) from that which will become the belly. In each case, the gradient thresholds may turn particular genes on and off in a series of elaborations that begins with the crudest determinants of shape – the front/back and top/bottom axes, say – and works its way to the fine details.
The idea that chemical concentration gradients might control the development of embryos was first proposed at the start of the twentieth century by Theodor Boveri. By producing a chemical patterning signal that spreads into the rest of the embryo, one cell can determine the fate of other cells nearby. In 1924 Hans Spemann, together with Hilde Mangold, called such groups of cells “organizers”.10 Mangold transplanted groups of cells in amphibian embryos from one position to another and saw that they could induce the development of “out of place” features.
The British biologists Julian Huxley and Gavin de Beer verified the idea of organizers in the 1930s by manipulating the embryos of birds. They proposed that Spemann’s organizers create “developmental fields” of some kind that influence the course of development. Spemann had imagined this “field” as something like the magnetic or electrical fields of physics, but Huxley, de Beer and their contemporaries in this fledgling field of developmental biology suspected that the agent was a chemical one. The notion that these organizing centres define a sense of position within the emerging body plan through the action of morphogen concentration gradients was crystallized in the late 1960s by biologist Lewis Wolpert.
There’s a crucial part of this story that I’ve skipped over so far. The patterning of the fruit-fly embryo is kicked off by the production of the bicoid protein at the anterior tip of the egg. But what causes that production in the first place? How does the bicoid gene know it is at the anterior end?
The answer is that “mother tells it”. While the unfertilized egg is attached to the follicle of the mother fly, specialized cells called nurse cells deposit material needed to make bicoid – specifically, the RNA molecules that mediate the gene-to-protein conversion – into the anterior tip of the egg, so that developmental patterning is all ready to go when the egg is fertilized. Right from the outset, cells in the embryo are dependent on other cells around it to know what to do. It’s for similar reasons that a fertilized human egg can’t develop fully in isolation, if cultured in a test-tube. Implantation in the uterus wall is needed to give it a “sense of up and down”. Ectopic pregancies (within the fallopian tubes) show that such a signal doesn’t have to come from the uterus, however, and we’ll see later whether there might be other ways to do produce it in vitro.
This is why it is strictly incorrect to say – although it often is said – that all the information needed to grow a human being is in the genome of the fertilized egg, which is in turn supplied by the gametes that combined to make it. You could say that the human embryo also needs positional information supplied by its environment – specifically by the uterus lining. Furthermore, any particular cell in the developing embryo depends on receiving information from the surrounding cells in order to keep embryo growth on track. As the transplantation experiments of Huxley and de Beer showed, if you mess with that information then you screw up development, despite the fact that every cell retains its complete “genetic programme”.
Embryo development is thus not encoded from the outset in the genome, as if in some blueprint or instruction book. It relies on a precise expression of genetic information in time and space, which in turn depends on the proper coordination of many cells (including maternal ones) and is subject to chance events during the execution. To understand embryology and the growth of complex tissues and organisms, we shouldn’t imagine that we will find a set of instructions packed like a homunculus inside the zygote. Rather, we will need to discern and interpret the patterns of information flow (and the various sources of that information) as the process unfolds.
It’s rather as if the genome is a list of the words that feature in a book, but you need other information to put the words in the right order so that they become more than just assemblages of letters and may take on meanings. Those meanings are not inherent in the words themselves but may be determined by the words nearby, by allusions and interactions that leap from one part of the text to another – by context. Again, there is no perfect metaphor for illustrating how genes work in building an organism; doubtless this one would collapse too under pressure, so use it gently.
* * *
I won’t explain in detail how human embryogenesis differs from that of the fruit fly, but it’s worth understanding one of the most basic distinctions. For the human body doesn’t simply emerge imprinted on the inner cell mass of the blastocyst like stripes on an embryonic zebra.11 Rather, the cells in the embryo move around, and the tissues grow, buckle and fold, to shape the body. It’s a highly active process, a kind of auto-origami happening in parallel with the appearance of distinctions between cell types. The first stage of this process, which for humans occurs around day 14 after fertilization (around the time that a pregnant woman might first notice a missing menstrual period), is called gastrulation. Some scientists regard this as the point where a mass of cells begins to produce an organism: as the beginning of personhood.
There is a lame joke that scientists still seem to find amusing about how, if a physicist were to study the cow, she would first simplify the question by approximating it as a sphere. It is rendered all the lamer by the fact that this is not so far from how nature approximates the human body – or the bovine one for that matter – in the first instance. For the most rudimentary idealization of our body might run thus: an inner tube for digestion from mouth to gut to anus, an outer layer of skin to create a boundary, and everything else packed into the space in between. At one end we’ll put the head – the anterior – and at the other end is of course the posterior. Gastrulation creates a structure very much like this (the word actually means “gut formation”). In some creatures, such as species of worms and molluscs, it really is that simple: gastrulation folds the embryo into a sort of fat tube or doughnut shape in which an inner tube connects mouth to anus, and the job is nearly done at a stroke.
For humans, it is rather more complicated. The embryo develops a central groove called the primitive streak, which will become the axis of the backbone and central nervous system: the beginning of the aforementioned neural tube. The subsequent folding is not easy to describe in words, but it creates the crescent-shaped structure that will become the fetus, connected to a yolk sac (involved in early embryonic blood supply) and attached to the placenta via the umbilical cord. The key point is that initially this gastrulated human embryo develops distinct types of tissue: its cells lose their pluripotency and start to specialize. The innermost layer, which will form the lining of the gut, is called the endoderm (“inner skin”). The outer layer, or ectoderm, generates the surface layer of the skin as well as the brain and nervous system. Between these layers is the mesoderm (“middle skin”), which is the primal fabric of the inner organs and tissues: the heart, kidneys, bone, muscles, ligaments and also the blood. At this stage, some of the embryonic stem cells are also set aside to become the germ cells: the precursors to the gametes (eggs and sperm).
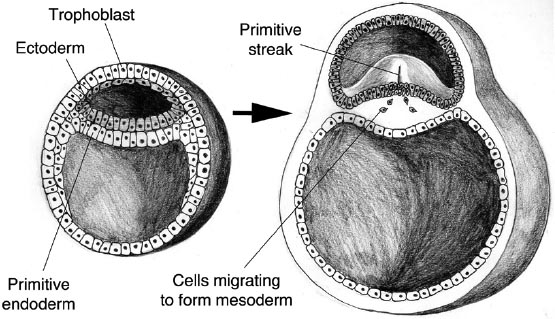
Gastrulation of the human embryo and formation of the primitive streak.
And there you have it: the schematic human body, its cells launched on their road to specialization. The rest is refinement. For example, some neural cells in the head region develop (around week five of gestation) not into neurons but into the retinal cells of the eye. Some cells don’t differentiate where they first sit, but actively migrate through the embryo to where they need to be – we saw that the primordial germ cells do this. The sex organs develop identically at first in both sexes, becoming female organs unless triggered into the structures of the male if the cells have a Y chromosome instead of a second X. On the Y chromosome sits a gene denoted SRY, which controls other genes needed to develop male characteristics.
All of this refinement happens through cell dialogue. Molecular messages pass from cell to cell, each at the proper stage of development, so that cells get assigned their roles in collaboration with their neighbours. “The parts of each organ help the other parts to form,” explains cell biologist Scott Gilbert. It is because organoids like my mini-brain lack this information from surrounding tissues that their development – their morphology – is imperfect. To make a body or even a mature organ, cells need community.
* * *
The idea that genes involved in development interact and control one another via diffusing morphogens is only a part of the story of how embryos take on their form. The distinctions between cell types initiated by such signals become permanently imprinted on the cells as they develop into different tissues.
How can that be, if they still all share the same genome?
That problem was recognized by Thomas Hunt Morgan and others in the early days of molecular genetics, but no one really knew how to address it. So it was largely put to one side. The discovery of DNA’s genetic code in the 1950s and ’60s all but eclipsed the question, seeming as it did to promise an underlying simplicity in the way cells function. Already in 1941, Morgan’s former student George Beadle, along with biochemist Edward Tatum, had shown that genes (whatever they were – it wasn’t yet clear) encode protein enzymes. This became understood to mean that each gene has a unique corresponding protein. The key question was then how a gene made a protein. Crick and Watson’s double helix, zipped together with information-bearing nucleotide bases, seemed to deliver the answer: DNA carries the coded plan, and RNA and ribosomes are the machinery that does the translation.
But how do you get from a protein to the phenotypic effect of a gene on the unfolding organism? That wasn’t at all obvious. By the 1960s, the general idea was that genes act in some vague way to dictate the developmental programme, which was then envisaged merely as “an unfolding of pre-existing instructions encoded in the nucleotide sequences of DNA”, as American biologist-cum-historian-cum-philosopher Evelyn Fox Keller has put it. According to the French biochemist Jacques Monod, as far as gene action is concerned, “what’s true for [the bacterium] E. coli is true for the elephant.” What seemed to matter was establishing the common basis by which gene becomes protein. Somehow the rest – meaning the living organism itself – followed. Which would be all very well, if E. coli looked like an elephant.
In this picture, then, the answer to the question of development must reside in the gene sequence, and the ultimate goal of biology becomes the decoding of that sequence. This picture has been burnished for a remarkably long time, culminating in the Human Genome Project, which began in the 1990s and announced the almost complete sequencing of the human genome between 2001 and 2003.12 The objective was simply to get the code, which took on the status of the “fundamental” information directing all biological activity. Meanwhile, genetics more broadly looked for correlations between genes and phenotypic outcomes. Exactly how and why genes exert their effects was a question long bundled up in the vague concept of “gene action” that, as Keller says, allowed scientists “to get on with their work despite almost complete ignorance of what that ‘action’ consisted of.” There was an implied hierarchy of causation in which genes were paramount, as reflected in Nobel laureate David Baltimore’s comment that the development of an organism involved the “greasy machines” of the cell directed by the “executive suite” of the genome. (Engineers are very familiar with this kind of prejudice.)
The resulting view was that development was a kind of painting by numbers of the plan in the genome. For a complex organism like us, that left an awful lot of instructions to be packed into the genes. As the Human Genome Project got underway, biologists estimated the number of genes the project would find as being somewhere between 140,000 and a lower limit, proposed by a few bold souls, of 26,000. Most put the figure at around fifty to seventy thousand.
The answer turned out to be 23,000.
This is often presented as a sobering example of how experts can get things wrong. It’s certainly that, but rarely does anyone identify the real moral: that the genome doesn’t work the way it was thought to.
Zoologist Fred Nijhout is one of the few to have come properly to terms with the implications. “A more balanced and useful view of the role of genes in development,” he says, “is that they act as suppliers of the material needs of development and … as context-dependent catalysts of cellular changes … they simply provide efficient ways of ensuring that the required materials are supplied at the right time and place.” They are less like Baltimore’s executive directors, and more like stewards guiding a crowd. It’s no coincidence that Nijhout sees things this way, because he is an expert on the genetics of butterfly wing patterns, where it is clear that just a few genes, creating interacting fields of influence through the diffusion and spreading of morphogens, can generate a startlingly diverse array of patterns and forms, dictated by the details of how the genes are expressed in time and space. It’s somewhat meaningless, in such a situation, to say what a gene does (beyond “encode a class of proteins”) without specifying where and when it acts.
The view now emerging is that a relatively small number of genes is able to generate the complexity of the human form, with its many different tissue types so precisely arranged and coordinated, because they act in networks that produce distinct patterns of gene expression varying over time. With 23,000 genes, the number of possible networks of influence is astronomical.
How do genes acquire and change their patterns of behaviour? The control, activation and silencing of genes in different cell types and at different stages of development is called epigenetics. The word literally implies something additional to genetics, but what it really connotes is that the observable outcome of genetic activity – the phenotype, such as the tissue type of a cell – isn’t determined by the genotype (that is, which genes are present), but by the question of which genes are active. Epigenetics is all about how genes become modified to alter whether, or how much, they are expressed.
There are several ways in which this can happen. One is by the attachment of molecular “tags” to the respective genes, which might act as markers that deter the machinery of transcription, suppressing gene expression. Some genes can be switched off, for example, by proteins that stick a so-called methyl group – a carbon atom with three hydrogen atoms attached – onto DNA bases in the gene, which forms a sort of “shield” that protects the gene against being transcribed and translated into a protein. Another molecular mechanism of epigenetic regulation involves chemical changes to the histone proteins around which a stretch of DNA is wrapped in the chromosomes.
Harder to understand than this attachment of “leave me alone” labels to genes, but equally important for epigenetics, are processes that involve the packaging of DNA in chromosomes. Remember that the combination of DNA and histone proteins in chromosomes goes by the name of chromatin. This stuff is rather systematically coiled up and stowed when the chromosomes are in the compact form (typically X-shaped) found in dividing cells. At other points in the cell cycle, chromatin can be unwoven and loosely strewn, in which case the transcription machinery can get to it more easily. So how “active” genes are can depend on how the corresponding regions of the chromosomes are packaged.
An example of this epigenetic gene regulation happens in female cells, which contain two copies of the X chromosome, one passed on from each parent. If both of them were active, they would produce more proteins from this chromosome than the cell needs, and that would cause problems. So one X chromosome is inactivated early in the development of the embryo. The choice of whether to silence the maternal or paternal X chromosome is made by each cell at random and then passed on to daughter cells when they divide. The result is that females end up with an equal blend of two types of cell throughout their body. This process of X-chromosome inactivation was first identified by geneticist Mary Lyon in the 1960s. It took many years to figure out how “X-silencing” occurs, but we now know that it involves a gene that switches on a series of events resulting in the packaging of the inactive chromosome into a tight bundle, inaccessible to transcription. All the genes are still there and are faithfully copied and passed on when cells divide, but the shape of the chromosome keeps them hidden.
Some epigenetic changes to DNA that regulate gene activity happen automatically as cells divide and mature: each cell type will have its own characteristic pattern of epigenetic modifications. This too is why development of a fertilized egg into an embryo and then a mature organism isn’t exactly just a matter of reading out a genetic programme. It involves a continual, ever-changing process of epigenetic editing of that programme, taking place through time and space.
* * *
In the mid-twentieth century, British embryologist Conrad Hal Waddington offered a metaphor for the process of epigenetic cell differentiation. He imagined cells in the early embryo traversing a landscape of possibilities: they begin their journey at a mountain peak and descend into valleys that branch like the channels of a river. At each branching point, the cell (more properly, the lineage of dividing cells) makes a decision about its subsequent fate: to become a progenitor of lung or heart, say. The consensus was that, once a lineage has descended into a valley, it can never reverse direction and go back uphill again.

The Waddington landscape. The balls represent the trajectories of different cell lineages, which begin in the single valley of totipotency as the zygote first begins to divide, and end in valleys representing different types of mature, differentiated cell.
Differentiation begins early. Indeed, it has happened even in the pluripotent embryonic stem cells of the epiblast, which have lost the totipotency of the earliest cells made from the dividing zygote. Already some of those first cells have been directed down the valley in Waddington’s landscape that leads to a placenta or a yolk sac, not to a part of the fetal body. The cells that make up the three layers of endoderm, mesoderm and ectoderm in the gastrulated embryo have undergone a further degree of differentiation, a further narrowing of choice.
It’s because of this specification of cell “fates” early in embryogenesis that the germ cells need to be formed so soon. Evidently a barely formed embryo doesn’t yet “need” eggs or sperm – but it must put aside the cells from which they will grow before they lose too much of their pluripotency. The germ cells, after all, have to make a totipotent zygote, so it won’t do if their chromosomes have already been heavily modified and silenced. Germ cells do have some epigenetic silencing of genes, although this too must be stripped away when the gametes combine to make a totipotent zygote.
This special dispensation for germ cells aside, epigenetic changes appear to be one-way. They partly account for how our body tissues remember and maintain their identity as they grow: why skin cells divide to produce more skin cells, and don’t spontaneously become muscle cells or stem cells. In other words, cell replication is somewhat more complex than merely a matter of copying the chromosomes. It’s necessary also to copy the epigenetic chromosomal modifications that give the cell its identity.
What this means is that each cell in our bodies, like each one of us, has a lineage: an ancestral history that starts with the zygote – and, except for a handful of germ cells (if we have children), ends in the grave. A liver cell has arisen from an embryonic stem cell via a succession of ancestors with intermediate characteristics, reflecting an ever greater specialization and loss of versatility. This notion of cell lineages was first articulated clearly by August Weismann when he drew up his fundamental distinction between somatic (“mortal”) and germ (“immortal”) cells.
When we tell the story this way, a new possibility becomes apparent. In cells during development, just as in organisms during evolution, genes can change. Every time a cell divides, there is a chance that some of the three billion base pairs in the genome will be miscopied – that the daughter cells will acquire mutations. Cells put a great deal of effort into avoiding such mistakes, employing a kind of molecular proofreading to check for errors in replication. All the same, the numbers are so vast that mutations are inevitable. It’s estimated that distributed within the chromosomes of our 37 trillion or so cells are about ten thousand trillion genomic mutations.13 Every one of our genes experiences somatic mutations at some point in our lives.
Most of these mutations, fortunately, don’t matter – they don’t affect a gene’s ability to do its job(s). But some have consequences. Most notoriously, gene mutations can lead to the cell dysfunctions that cause cancer (see here). Even potentially harmful mutations, though, might not matter if they happen late in development and so appear only in a few cells. Somatic mutations that arise during early embryo growth, on the other hand, may be passed on to all subsequent cells in that lineage, making the developing body a patchwork or “mosaic” with slightly but perhaps significantly different genomes. There are many diseases related to “mosaicism”, of which cancer is just one class. Somatic mutations leading to mosaicism are particularly common in brain neurons, and they are thought to be responsible for a range of brain and cognitive disorders, including some types of autism. Even benign mutations can manifest themselves in outward appearance (that is, phenotypically): for example, causing striped skin pigmentation called Blaschko’s lines or the red skin blotches called port-wine stains.


