


Полная версия
Боковой амиотрофический склероз
Существует целый ряд хорошо охарактеризованных трофических факторов для ЦНС, таких как нейротрофический фактор головного мозга (BDNF), инсулиноподобный фактор роста 1 (IGF1), цилиарный нейротрофический фактор (CNTF), глиальный нейротрофический фактор (GDNF), фактор нервов (NGF), гормон роста и васкулярный эндотелиальный фактор роста (VEGF). Многие из них тестировались на наличие нейропротекторных свойств в различных экспериментальных моделях БАС. В действительности вирусные векторы, кодирующие факторы роста, являются одним из наиболее эффективных способов сдерживания прогрессирования дегенеративных процессов и пролонгации выживания мышей с БАС (Wang et al., 2002; Kaspar et al., 2003; Azzouz et al., 2004; Dodge et al., 2008).
Недостаток специфических трофических факторов приводит к нарушению дифференцировки двигательных нейронов, а отсутствие какого-либо трофического сигнала приводит к нарушению развития разнообразных субпопуляций двигательных нейронов. Отсутствие глиального нейротрофического фактора видоизменяет местоположение развивающихся двигательных нейронов, иннервирующих конечности (Haase et al., 2002; Kramer et al., 2006), а также выборочно подавляет иннервацию интрафузальных мышечных веретен (Gould et al., 2008). Любопытен факт, что чрезмерная экспрессия данного фактора в мышцах при их развитии вызывает гипериннервацию нейромышечных соединений (Nguyen et al., 1998). В противоположность этому нейротрофический фактор головного мозга может и не оказывать большого влияния на двигательные нейроны, так как, несмотря на то что недостаток этого фактора оказывает существенное негативное воздействие на нормальное развитие сенсорных нейронов, двигательные нейроны способны развиваться без крупных изменений (Ernfors et al., 1994a; Jones et al., 1994). Более того, отдельные субпопуляции двигательных нейронов обнаруживают признаки различной степени чувствительности к недостатку нейротрофинов. Так, отсутствие нейротрофина-3 приводит к полной потере спинальных двигательных нейронов (Ernfors et al., 1994b; Gould et al., 2008), тогда как двигательные нейроны лица остаются незатронутыми, а отсутствие цилиарного нейротрофического фактора CNTF не вызывает изменений в развитии двигательных нейронов на спинальном или краниальном уровнях (DeChiara et al., 1995), хотя потеря рецептора α этого фактора (CNTFRα) приводит к значительному дефициту моторных нейронов, и мыши, испытывающие недостаток в этом рецепторе, погибают в перинатальном периоде (DeChiara et al., 1995). Возможным альтернативным или дополнительным лигандом для этого рецептора может служить димер, образованный кардиотрофиноподобным цитокин/цитокиноподобным фактором 1, истощение которого способно вызывать существенное снижение количества двигательных нейронов (Forger et al., 2003). Также было обнаружено, что отсутствие иных факторов, таких как кардиотрофин-1, вызывает существенную потерю двигательных нейронов (Oppenheim et al., 2001; Forger et al., 2003), а отсутствие инсулиноподобного фактора роста-1 (IGF1) вызывает значительное сокращение численности тригеминальных и лицевых двигательных нейронов (Vicario-Abejón et al., 2004). Наконец, в то время как недостаток фактора эндотелиального роста сосудов (VEGF) носит летальный характер, истощение элемента гипоксической ответной реакции в промотерном отделе гена VEGF вызывает снижение экспрессии этого фактора, что приводит к приобретенной прогрессирующей потере двигательных нейронов в мышиных моделях (Oosthuyse et al., 2001). После этого открытия стало известно, что определенные VEGF гаплотипы (-2578С/A, -1154G/A и -634G/C) передавали повышенную восприимчивость к БАС в работах с людьми, но позднее, при проведении метаанализа с участием более 7000 испытуемых, собранных из как минимум 8 различных популяций, не было выявлено какой-либо взаимосвязи между этими гаплотипами и возникновением БАС (Lambrechts et al., 2009). Кроме того, у больных с БАС не были выявлены мутации ни в элементе гипоксической ответной реакции промотерного отдела VEGF (Gros-Louis et al., 2003), ни в VEGF рецепторе 2 (Brockington et al., 2007).
Нейротрофические факторы оказывают большое воздействие не только на ход развития, они также управляют сохранением и поддержанием, выживанием двигательных нейронов на протяжении длительного времени даже после дифференцировки. Таким образом, они могут запускать активирование эндогенных регенеративных процессов. Помимо синтеза трофических факторов в локальной спинальной микросреде, синаптические мишени двигательных нейронов также играют важную роль в механизмах трофической обратной связи. На самом деле этот факт представляется первостепенным для развития ЦНС, так как при этом трофические элементы тканей мишеней достигают формирующиеся нейроны, что позволяет им преодолевать эндогенно закодированную гибель клеток (Oppenheim, 1991). В случае с двигательными нейронами подобные эффекты преимущественно достигаются при помощи факторов, происходящих из скелетных мышц (Oppenheim et al., 1988; Grieshammer et al., 1998; Kablar and Rudnicki, 1999).
Среди всех трофических факторов, протестированных в экспериментальных моделях БАС, фактор VEGF оказался одним из наиболее действенных защитников двигательных нейронов. Фактор VEGF замечательным образом замедляет прогрессирование заболевания и отмирание двигательных нейронов в семейных (Azzouz et al., 2004; Zheng et al., 2004; Storkebaum et al., 2005; Wang et al., 2007), а также спорадических (Tovar-Y-Romo et al., 2007; Tovar-Y-Romo, Tapia, 2010, 2012) опытных моделях моторной нейродегенерации. Активация VEGF рецептора 2 запускает фосфорилирование внутриклеточных путей, управляемых фосфатидилинозитол-3-киназой (PI3-K), фосфолипазой C-γ и митоген-активированной протеинкиназой (MEK), которые способствуют ингибированию проапоптозных факторов, в том числе Bad (Yu et al., 2005), каспазы 9 (Cardone et al., 1998) и каспазы 3 (Góra-Kupilas, Joško, 2005; Kilic et al., 2006). Активация подобных внутриклеточных сигнальных путей была тщательно изучена применительно к ЦНС (Zachary, 2005). VEGF-зависимая активация PI3-K/Akt считается достаточной для предотвращения моторной нейрональной гибели в семейных моделях БАС in vitro (Li et al., 2003; Koh et al., 2005; Tolosa et al., 2008), а также в опытных моделях эксайтотоксической нейрональной гибели in vitro (Matsuzaki et al., 2001). Более того, активация PI3-K/Akt требуется для выживания двигательных нейронов и аксональной регенерации после спинномозгового поражения (Namikawa et al., 2000). Было доказано, что сигналинг, опосредованный PI3-K, критическим образом связан с защитным воздействием фактора VEGF против AMPA-индуцированной эксайтотоксической спинальной нейродегенерации в условиях in vivo (Tovar-Y-Romo, Tapia, 2010).
VEGF также выступает посредником в нейропротекции, осуществляемой через ингибирование стресс-активируемых протеинкиназ, в том числе p38 митоген-активируемой протеинкиназы. Повышенные уровни фосфорилированной p38 отмечались в моторных нейронах и в глии в моделях семейных случаев БАС на мышах (Tortarolo et al., 2003; Holasek et al., 2005; Veglianese et al., 2006; Dewil et al., 2007), даже в пресимптоматической стадии (Tortarolo et al., 2003). А также p38 является важным фактором для пути клеточной гибели, специфическим для двигательных нейронов (Raoul et al., 2006). Заслуживает внимания то, что ингибирование p38 может предотвращать гибель двигательных нейронов в in vitro семейной модели БАС (Dewil et al., 2007). Несколько групп исследователей доказали, что VEGF может подавлять активацию p38 как в семейных, так и в эксайтотоксичных моделях (Tovar-Y-Romo, Tapia, 2010) спинномозговой нейродегенерации.
Повышенная экспрессия VEGF-индуцирующего гипоксия-индуцированного фактора 1 (HIF-1α), наблюдаемая в спинном мозге, может быть вызвана взаимосвязанными гипоксическими состояниями спинальной микросреды, хотя моторные нейроны не кажутся способными к полномасштабному ответу на повышенный уровень нижележащих эффекторов, таких как VEGF (Sato et al., 2012). Одним из возможных объяснений этого обстоятельства, а также снижения экспрессии VEGF, наблюдаемой у пациентов (Devos et al., 2004), может быть тот факт, что индуцирующие факторы, такие как HIF-1α, не допускаются и исключены из процесса перемещения в ядро, несмотря на то что их концентрации увеличиваются в цитоплазме (Nagara et al., 2013). Подобная неспособность задействовать полную ответную реакцию синтеза VEGF во время гипоксии не носит специфический характер для клеточных типов, и такая неспособность отмечалась в моноцитах, полученных у больных БАС (Moreau et al., 2011).
В отличие от хорошего защитного потенциала фактора VEGF, другие факторы, в том числе BDNF, не оказывали защитное воздействие в разных экспериментальных условиях. Фактор BDNF синтезируется посредством активированной микроглии на ранних стадиях заболевания, когда глиальная ответная реакция преимущественно оказывает противовоспалительное и защитное воздействие, однако выработка этого фактора утрачивается при приобретении микроглией токсических элементов на более поздних стадиях (Liao et al., 2012). Кроме того, BDNF не защищает двигательные нейроны от эксайтотоксичности в экспериментальных моделях in vitro (Fryer et al., 2000) и in vivo (Tovar-Y-Romo, Tapia, 2012). Возможно, это объясняется секвестрацией лиганда, которая происходит за счет усеченной изоформы рецептора высокой аффинности, о котором известно, что он выделяется двигательными нейронами, поскольку устранение этого усеченного рецептора значительно замедляет начало болезни в семейной модели на мышах (Yanpallewar et al., 2012). Несмотря на это, BDNF может оказаться фактором риска для нейронов, либо ввиду того, что он увеличивает их чувствительность к эксайтотоксичности (Fryer et al., 2000), либо за счет механизма активации NADPH оксидазы – фермента (Kim et al., 2002), участвующего в патологии двигательных нейронов посредством разрушения путей выживания, активируемых трофическими факторами (Wu et al., 2006). Было показано, что другие факторы роста также оказывали благотворное воздействие, хотя и в меньшей степени.
Экспрессия фактора GDNF, осуществляемая астроцитами, повышается на фоне спинномозговой ишемии, и это может служить механизмом защиты по отношению к моторным нейронам против эксайтотоксической гибели (Tokumine et al., 2003). Фактор GDNF оказывает свое нейропротекторное воздействие преимущественно на нейрональные сомы, нежели на нервные окончания в нейромышечных синапсах, будучи задействованным непосредственно в спинном мозге (Suzuki et al., 2007). Напротив, при воздействии непосредственно на мышцы фактор GDNF сохраняет мышечно-нервный синапс и стимулирует функцию моторных нейронов и их выживание в семейной модели БАС (Suzuki et al., 2007), что дает основание предположить, что защитные свойства, осуществляемые фактором GDNF, носят довольно ограниченный характер и распространяются в пределах трофического источника. Тем не менее GDNF может ретроградно транспортироваться вдоль моторных нейрональных аксонов (Leitner et al., 1999), что создает условия для изучения маршрута доставки, который мог бы влиять как на сомы, так и на нервные окончания. Любопытно, что у людей с БАС выявляются признаки повышенной экспрессии фактора GDNF в мышцах (Grundström et al., 1999), а сверхэкспрессия данного фактора в мышцах, но не в астроцитах приводит к увеличению продолжительности жизни у мышей с моделью БАС (Mohajeri et al., 1999). Комбинированная терапия с использованием факторов роста может оказаться альтернативой, заслуживающей должного изучения, о чем свидетельствует недавнее сообщение о трансгенной модели БАС на крысах, на которой было показано, что факторы VEGF и GDNF, вводимые через имплантат мезенхимальных стволовых клеток человека, оказывают синергическую защиту нервных синапсов мышц (Krakora et al., 2013).
Что касается фактора CNTF, хотя и сообщалось, что блокировка его экспрессии приводит к потере моторных нейронов (Masu et al., 1993) и развитию двигательных симптомов, подобное воздействие носит сравнительно легкий характер в сравнении с действием, индуцируемым потерей других факторов, в частности VEGF. Интересно также то, что у больных БАС выявляется избирательное снижение экспрессии CNTF в тех отделах ЦНС, которые подверглись негативному воздействию болезни (Anand et al., 1995). Напротив, сывороточные уровни CNTF обычно повышены у больных БАС, особенно у пациентов с формой люмбального начала заболевания (Laaksovirta et al., 2008).
Глава 3. Генетические аспекты бокового амиотрофического склероза
В настоящее время доминирует генетическая теория происхождения БАС. Приблизительно 5—10% случаев заболевания являются семейным БАС (FALS), остальные 90—95% случаев – спорадические БАС (SALS). Известно, что около 20% семейного и 5—7% спорадического БАС связаны с мутациями в гене медь-цинк зависимой супероксиддисмутазы (СОД-1) – фермента, утилизирующего свободные радикалы.

Рис. 3. Единственный каузативный ген, кодирующий супероксиддисмутазу-1 (СОД-1), мутации которого приводят к БАС
К настоящему времени открыто 108 мутаций генов при боковом амиотрофическом склерозе. Все, кроме D90A и D96N, наследуются по аутосомно-доминантному типу. Помимо этого, за последнее десятилетие выявлено несколько генов помимо гена СОД-1, мутации которых могут приводить к развитию БАС: гены белков цитоскелета мотонейрона, гены белков, регулирующих выживание мотонейронов, гены белков митохондриальной дыхательной цепи. Основные описанные генетические локусы при боковом амиотрофическом склерозе представлены в таблице 1 и 2.
На данный момент определены более 22 локусов БАС и 4 локуса БАС + FTD (FTD-ALS – frontotemporal dementia – ALS) (БАС наряду с лобно-височной деменцией), и в большинстве случаев идентифицируются гены, связываемые с возникновением заболевания. Это обозрение подводит итог доступной на сегодня информации, которую удалось собрать по четырем наиболее распространенным генам, связываемым с семейными случаями БАС: SOD1, TARDPB, FUS, и C90RF72. Эти гены привлекли внимание к роли окислительного стресса и РНК-процессинга как патогенных механизмов, способствующих развитию БАС.
⠀
Таблица 1.
Описанные генетические локусы при боковом амиотрофическом склерозе, по данным отечественной литературы
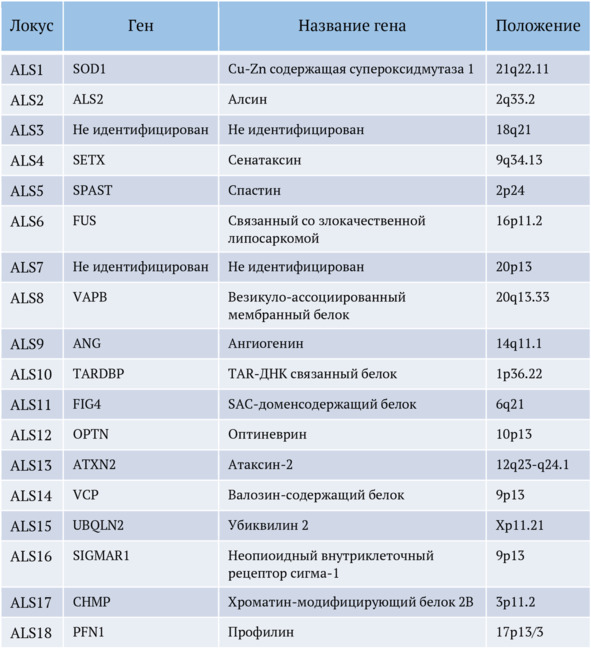
Кроме того, более редкие генетические аллели заключают в себе дополнительные биологические пути, как, например, систему убиквитин-протеасомы (UPS), белковый трафик, а также расстройство цитоскелетной функции.
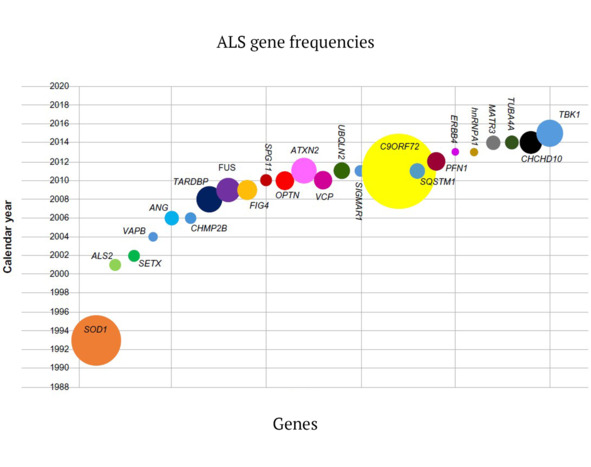
Рис. 4. Исторические вехи и частота открытия различных генов, участвующих в возникновении бокового амиотрофического склероза. Примечание. Каждый ген нанесен на график в соответствии с годом его обнаружения. Размер мутаций в FALS и ALS, как указано в литературе. В тех случаях, когда частоты генов отсутствуют, для иллюстративных целей им присваивается размер круга, эквивалентный 1% (цит. по: Al Sultan et al., 2016)
На представленном рисунке 4 авторы использовали нумерацию локусов, как это приведено в Online Mendelian Inheritance (онлайн каталог фенотипических маркеров у человека) в каталоге фенотипических серий по БАС (PS105400) и FTDALS (PS105550) (Online Mendelian Inheritance in Man, OMIM®, 2016).
БАС1: Cu-Zn дисмутаза супероксида (SOD1). Мутация Cu-Zn супероксиддисмутазы 1 (SOD1) была первой описанной генетической причиной семейных случаев БАС (FALS) (Rosen et al., 1993). Большинство SOD1-мутаций представляют собой аутосомно-доминантную картину, сопровождаемую высокопроникающим паттерном наследственности. Эти мутации преимущественно ассоциируются с возникновением БАС в каких-либо конечностях. Исключением из данного правила является мутация D90A, выявляемая преимущественно у скандинавских популяций, в которых она наследуется в аутосомно-рецессивном виде. Частота SOD1-мутаций варьируется в зависимости от популяций, от 23% в Скандинавии и до 12% в Германии; также мутации определялись в кажущихся спорадических случаях (Andersen, 2006). База данных по БАС (ALSoD database, http://alsod.oip.kcl.ac.uk; Abel et al., 2012) сообщает о 183 мутациях в SOD1, связываемых с болезнью (оценка на ноябрь 2015 г.), большая часть которых представляет собой точечные мутации.
Учитывая, что SOD1 кодирует 153-й аминокислотный белок, это количество мутаций поразительно, причем мутации распределяются по всему гену и оказывают воздействие на целый ряд доменов внутри белка. Данное обстоятельство отличается от некоторых других ассоциированных с БАС мутаций, которые чаще локализуются в пределах определенного мотива выделяемого белка, тем более что неясно, являются ли все заявленные SOD1-мутации действительно патогенными (Felbecker et al., 2010; Marangi, Traynor, 2015). Множественные мутации, происходящие внутри белка, привели также к трудностям определения того, каким образом они отвечают за фенотип болезни. Ген SOD1 является повсеместно экспрессируемым антиоксидантным белком, ускоряющим и катализирующим супероксид свободных радикалов в перекись водорода и кислород. Так как большинство мутантных белков сохраняют эту ферментативную функцию, предполагается, что патогенность оказывает свое воздействие через принцип токсического приобретения функции, хотя точная природа подобной токсичности пока еще остается полностью не изученной. Рассматривался ряд взаимно совместимых патогенных механизмов, в том числе окислительный стресс, экскайтотоксичность, скопление и агрегация белков, нейровоспаление, апоптоз, митохондриальная дисфункция, дисрегуляция аксонального транспорта и стресс эндоплазматического ретикулума (Felbecker et al., 2010; Marangi, Traynor, 2015; Kaur et al., 2016). Мутантные SOD1-белки (mtSOD1) обнаруживают переменные состояния металляции (замещения металлом водорода) и формирования дисульфидной связи, что приводит к тому, что деметаллизированная и развернутая апоформа может проникать в межмембранное пространство митохондрии (пластосомы), вызывая тем самым митохондриальную дисфункцию (cSheng et al1., 2012). Помимо этого, процесс деметаллизации приводит к повышению неустойчивости, и mtSOD1 демонстрируют более высокую предрасположенность к накоплению, чем SOD1 дикого типа.
Таблица 2. Обзор ключевой информации, доступной для локусов ALS и FTDALS, о генах, вовлеченных в развитие бокового амиотрофического склероза (цит. по: Al Sultan et al., 2016)
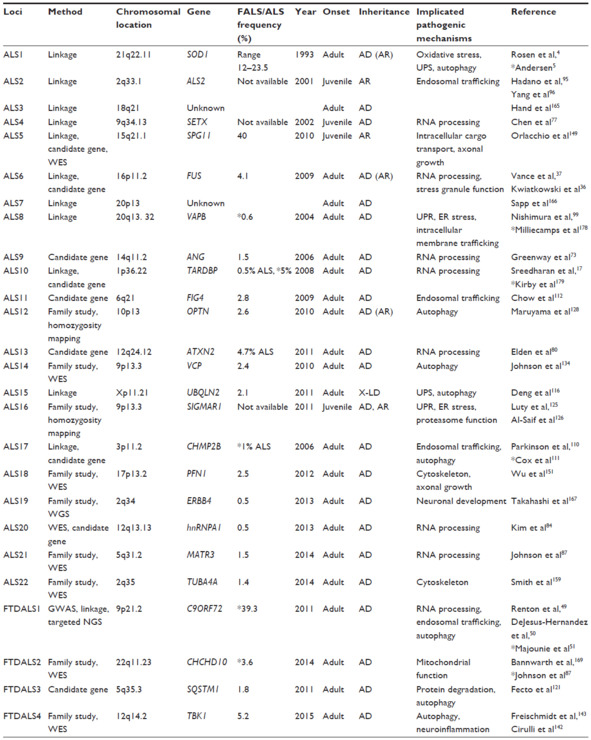
Примечание. Нумерация локусов ALS и FTDALS определяется онлайн-каталогом фенотипических маркеров у человека OMIM®. Частота мутаций в FALS основана на данных из исходных статей или ссылок, помеченных звездочкой (*). В некоторых случаях предоставляется частота в ALS, а не FALS (цит. по: Al Sultan et al., 2016).
Совсем недавно было показано, что мутантный SOD1 (mtSOD1), вместе с неправильно свернутым SOD1 дикого типа, продвигаются от клетки к клетке и инициируют прионо-подобное скопление SOD1 (Grad et al., 2014; Munch, Bertolotti, 2011). Тогда как первоначальное исследование продемонстрировало разрастание неправильно свернутого белка в моделях клеточных культур, спинальные гомогенаты, изъятые из парализованного мышиного мутанта G93A SOD1 и пересаженные в 6-месячную G85R-SOD1:YFP мышиную модель (в которой обычно болезнь не развивалась раньше, чем до 20 мес.), вызывали прогрессирующую болезнь двигательного нейрона в течение 3 месяцев (Ayers et al., 2016).
В обычном виде неправильно свернутые белки изымаются из клеток через механизм действия системы убиквитин-протеасомы (UPS). Однако в БАСе, обусловленном геном SOD1, а также в спорадическом БАСе было показано, что действие системы убиквитин-протеосомы (UPS) нарушается (Kabashi et al., 2012, Kabashi, Durham, 2006). Кроме того, было показано, что MGRN1 (mahogunin ring finger 1), являющийся E3 убиквитин-лигазой, которая катализирует и ускоряет моноубиквитинирование белков и помечает их для деградации при помощи UPS-независимого механизма, сокращается в G93A мышиной модели. Любопытно, однако, что сверхэкспрессия этого белка приводила к сокращению SOD1-токсичности за счет подавления агрегации (скопления) SOD1. Таким образом, терапевтические стратегии по лечению БАС включают повышение очищения от неправильно свернутого SOD1, и индуктор белка теплового шока, arimoclomol, представляет собой один из подобных препаратов, разрабатываемых и изучаемых в настоящее время (Kalmar et al., 2014).
Хотя вначале окислительный стресс считался одним из основных механизмов мутантных SOD1, продолжающиеся исследования патогенного действия SOD1 привлекли и такие факторы, как система убиквитин-протеасомы (UPS), накопление и деградация белков, а также другие аспекты белкового трафика. Подобные пути также связаны с открытием дополнительных генов семейных БАС (FALS) (Раздел «Белковый трафик и гены, имеющие отношение к деградации»).
ALS10/БАС10: TAR ДНК-соединительный белок (TARDBP)
Транзактивный ДНК-связывающий белок 43 (TDP-43) кодируется геном TARDBP на chr1p36.22 (Sreedharan et al., 2008). Ген TARDBP ответственен за 4—5% семейных БАС и примерно 1% спорадических случаев БАС (Millecamps et al., 2010). Мутации в TARDBP наследуются в виде аутосомно-доминантных (AD) проявлений и ассоциируются с классическим клиническим фенотипом БАС. Ген TARDBP кодирует несколько изоформ, из которых преобладает TDP-43. Изоформа TDP-43 является гетерогенным ядерным рибонуклеопротеином (hnRNP), наделенным сигналом ядерной локализации (NLS) и сигналом ядерного экспорта, что способствует челночному перемещению белка между ядром и цитоплазмой. В белке TDP-43 содержатся три последующих домена, два мотива распознавания РНК (RRM1 and RRM2), которые участвуют в соединении РНК и ДНК, а также глицин-обогащенный домен, который необходим для взаимодействия с другими белками и представляет собой локализацию, в которой происходит большинство мутаций (Baralle et al., 2013; Lagier-Tourenne et al., 2010).
Первоначально TDP-43 был идентифицирован как транскрипционный репрессор, соединяющийся с TAR ДНК в вирусе-1 иммунодефицита человека (Ling et al., 2015). После этого было продемонстрировано, что TDP-43 играет роль в РНК-метаболизме, в том числе в РНК-транскрипции, альтернативном сплайсинге, предварительном микроРНК-процессинге, РНК-транспорте и устойчивости матричной РНК (messenger RNA, mRNA) (Scotter et al., 2015). Белок TDP-43 обладает способностью самостоятельно регулировать экспрессию собственных генов за счет соединения с 3́нетранслируемой областью (3́UTR) своего матричного РНК, что порождает неустойчивость и разложение (Ayala et al., 2015). Также TDP-43 соединяется с UG-обогащенными последовательностями в многочисленных последовательностях mRNA (матричной РНК) в целях регулирования сплайсинга (Polymenidou et al., 2011; Sephton et al., 2011; Xiao et al., 2011). Кроме того, недавно была обнаружена новая функция, при которой TDP-43 может подавлять сплайсинг несохраненных (неконсервированных) экзонов, известных как криптические экзоны (Ling et al., 2015). Устранение TDP-43 позволяло этим криптическим экзонам встраиваться в последовательности mRNA (матричной РНК), что затем прерывало транслирование и вызывало нонсенс-опосредованное разрушение. Наконец, TDP-43 также известен в качестве составляющего компонента стрессовых гранул (SGs), хотя и неясно, способствует ли это обстоятельство процессу нейродегенерации (Aulas, Vande Velde, 2015). Роль белка TDP-43 весьма заметна в характерных убиквитиновых цитоплазматических включениях, которые обнаруживаются у больных с БАС и лобно-височной деменцией (Neumann et al., 2006). Примерно 97% больных с семейной и спорадической формами БАС являются положительными для TDP-43 включений в двигательном кортексе и спинном мозге, тем самым подчеркивается значение TDP-43 как основной белковой сигнатуры заболевания, а не только тех белков, переносящих TARDBP-мутации (Sreedharan et al., 2008; Qin et al., 2014).
Потеря ядерной локализации TDP-43 при БАСе хорошо задокументирована, и существуют данные о наступающем в результате этого дефиците сплайсинга в клеточных и животных моделях БАС, а также в образцах, взятых у пациентов (Ling et al., 2015; Highley et al., 2014; De Conti et al., 2015). Кроме утраты ядерной функции, цитоплазматическое приобретение функции также может способствовать нейродегенерации. Модель мыши с мутацией в сигнале ядерной локализации TARDBP человека, что ограничивало TDP-43 до уровня цитоплазмы, продемонстрировала увеличенную экспрессию связанных с транскрипцией и хроматиновой сборкой генов и генов обработки гестона 3́UTR (Amlie-Wolf et al., 2015). Важно отметить, что подобные транскрипционные изменения не наблюдались при добавлении антисмыслового олигомера к нокдаун TDP-43 экспрессии, что, таким образом, поддерживало идею о цитоплазматическом токсическом приобретении функции. Наконец, как и в случае прионоподобного распространения заболевания, описанного при SOD-БАСе, были также получены доказательства того, что TDP-43 олигомеры дикого типа могут распространяться горизонтально от клетки к клетке через микровезикулы, в том числе через лизаты головного мозга больных БАС, а также вертикально по аксонам (Feiler et al., 2015). Таким образом, снижение скоплений подобных мутантных протеинов постепенно становится все более широко используемой терапевтической стратегией.