


Полная версия
Боковой амиотрофический склероз
Д. Марфунин (2012) в своей исследовательской работе «О боковом амиотрофическом склерозе» обсуждает варианты сочетаний вирусного начала и сосудистых нарушений при БАС. Он абсолютно верно утверждает, что верхние моторные нейроны расположены в двигательной области коры головного мозга, то есть в прецентральной извилине. Мышцы тела имеют определенную проекцию на этой извилине. Известно также, что эта извилина получает артериальную кровь из двух артерий – передней мозговой и средней мозговой. Граница зон кровоснабжения передней и средней артерии лежит приблизительно между проекцией тела и головы. Как считает Д. Марфунин, соблазнительно предположить, что поражающий агент (если такой существует) мог проникнуть в извилину извне. Учитывая вышеописанную картину прогрессии болезни, можно предполагать, что этот агент должен быть живым, способным к самовоспроизводству и мог бы лежать в основе и быть причиной болезни. Но попытки обнаружить вирусные частицы и молекулы в пределах моторных нейронов, корковых или спинальных, или в пределах тканей скелетных мышц были безуспешными. Более того, при рассмотрении пула моторных нейронов пациентов БАС в пределах пораженной области обнаруживается лишь недостаточность нейронов, но нет никакого намека на какую-либо борьбу нейрона против предполагаемого внедрившегося агента, способного его разрушить, только умеренная воспалительная реакция в окружающих тканях, главным образом основанная на наличии реактивной микроглии. Если все же предположить, что такой агент существует, то возникает несколько вопросов. Во-первых, кто он? Во-вторых, почему поражает лишь моторные нейроны? Почему поражает весь моторный тракт от терминальной пластинки до верхних моторных нейронов? Почему этот тракт поражается не одновременно, а начинается с терминальной пластинки и далее процесс распространяется вверх, создавая картину «отмирания»? И, наконец, почему моторные нейроны, не сопротивляясь, запускают процесс апоптоза? Если в качестве ответа на один из этих вопросов предположить, забегая вперед, что моторный тракт поражается с целью (если таковая существует) достижения гипокинезии, то для этого достаточно разрушить терминальную пластинку или, в крайнем случае, аксон переднего рога. Зачем же поражается весь тракт, вплоть до клеток Беца? Реального ответа нет. Ни вирусная, ни сосудистая теории не дают ответа на эти вопросы.
Другую причину возникновения БАС предлагает посттравматическая теория. Изучив анамнез более 60 пациентов с БАС, а также 20 пациентов с БАС, имеющих объективно доказанные патологические извитости магистральных артерий шеи и артерий, кровоснабжающих спинной мозг, мы обратили внимание на наличие в анамнезе этих больных серьезной закрытой травмы позвоночника или головного мозга. Этот факт подтвердился почти у половины этих больных. В трех случаях возникновение болезни отмечалось через 6—8 месяцев после минно-взрывных повреждений (боевая травма, криминальная травма при взрыве в автомобиле и т.д.), в 10 случаях удавалось выявить тяжелые повреждения позвоночника при профессиональной спортивной травме или занятиях экстремальными видами спорта: горные лыжи, единоборства, падение с высоты, падение на мотоцикле, неудачное падение спиной при прыжках с парашютом и т. д. Однако убедительных подтверждений этого феномена мы не отметили, моделирование этого феномена на экспериментальных животных не приводило к развитию клинической картины болезни БАС (Брюховецкий и др., 1998). Возможно, модель травмы спинного мозга на крысах не может служить объективной моделью хронического сосудистого фактора в возникновении БАС у человека из-за серьезных отличий анатомии сосудов и особенностей, связанных с прямохождением человека. Необходимо смоделировать эту нозологию на обезьянах и окончательно отказаться от этой теории или доказать ее состоятельность.
Существует и другая точка зрения, объясняющая возникновение БАС у спортсменов. Это теория чрезмерной физической активности. Согласно этой гипотезе, риск заболеть БАС у профессиональных футболистов в 6,5 раза выше, чем у обычного населения (Chio et al., 2005). A. Al-Chalabi, P.N. Leigh (2005) показали, что травмы, полученные при игре в футбол, являются причиной возникновения БАС у целого ряда профессиональных игроков. Напомним, что в США болезнь впервые была описана у игрока в американский футбол Лу Герига (болезнь Герига). Это связано с рядом специфических факторов: 1) с физической активностью, независимо от вида спорта; 2) с микротравмами или спецификой физических упражнений; 3) с употреблением допинга; 4) с типичными для футбола экологическими факторами; 5) с генетическими факторами, которые связаны с чрезмерной физической работоспособностью.
Есть научно обоснованная гипотеза возникновения БАС как паранеопластического процесса. Описаны случаи сочетания БАС с раком легкого, раком молочной железы, раком щитовидной железы, раком кишечника, инсулиномой и даже с мультиформной глиобластомой. Однако в последующем было доказано, что во всех этих случаях БАС был самостоятельным заболеванием. Наши собственные исследования состояния иммунной системы при различных типах рака и при БАС показали, что в основе этих заболеваний лежит хроническая или острая иммунная недостаточность, которая обусловлена разноуровневыми и разнонаправленными мультимаркерными изменениями протеомного профиля клеточной поверхности гемопоэтических стволовых клеток (ГСК) пациента. Учитывая, что ГСК у каждого человека являются основным источником образования всех типов клеток его иммунной системы, глобальные отличия протеомного профиля этих клеток между собой позволяют утверждать, что эти заболевания имеют различные нозоспецифичные иммунные нарушения. Профиль маркеров белков клеточной поверхности ГСК при раке, у больных с БАС и у здоровых доноров костного мозга имеют фундаментальные отличия между собой, и они были картированы и профилированы нами и представлены в главе 8 этой книги.
Существует также теория влияния экзогенных факторов на возникновение болезни. Экзогенными этиологическими факторами являются специфические воздействия окружающей природы. БАС даже получил название «болезнь Гуам», в которой сочетается клиника бокового амиотрофического склероза (БАС), паркинсонизма и деменции (комплекс Гуам) – редкая эндемическая патология, в основе которой лежит прогрессирующая генерализованная дегенерация нейронов центральной нервной системы (Morris et al., 2004; Скворцова и др., 2005; Яхно, 2005; Завалишин, 2009). Впервые это заболевание было описано D.R. Koerner в 1952 г. у коренных жителей чаморро на острове Гуам в Марианском архипелаге, расположенном в восточной части Тихоокеанского бассейна. Были подробно изучены воздействия ряда токсинов в природной экосистеме на острове Гуам. По данным P.A. Cox et al. (Сox et al., 2003), S.J. Murch et al. (Мurch et al., 2004), одним из природных небелковых веществ, обладающих нейротоксическим действием, является beta-Methylamino-L-alanin. Это вещество синтезируют цианобактерии, расположенные на кораллах, которыми, в свою очередь, питаются морские черепахи. Частое употребление местными жителями мяса черепах приводит к повышению его концентрации в тканях в 10—240 раз. Таким образом, в пищевой цепи на острове Гуам формируется эндогенный нейротоксический резервуар, оказывающий влияние на метаболизм белков, что способствует развитию заболевания. По мнению S. Murch et al., этот механизм приводит к развитию болезни даже через несколько лет у чаморро, покинувших Гуам (Мurch et al., 2004).
D.R. Koerner (1954) впервые обратил внимание на необычное сочетание неврологических синдромов, частота которых в 50—100 раз превышала распространенность бокового амиотрофического склероза в других странах мира, который нередко носил семейный характер (Koerner, 1952). Позднее эта патология была описана в этом же регионе на полуострове Кии в Японии и в западной части Новой Гвинеи (Hermosura et al., 2009; Kokubo, Kuzuhara, 2003). Заболевание чаще встречается у мужчин (соотношение муж.: жен. – 1,7:1) в широком возрастном диапазоне – от 30 и свыше 70 лет (Morris et al., 2001). Максимальная частота приходится на возраст 55—64 года. Болезнь чаще имеет быстрый и фатальный характер. В качестве спорадических случаев БАС и фронтотемпоральная деменция описаны в других странах мира (Егоркина, Гапонов, 2007; Dickson, 2008; Hasegawa et al., 2007). Патогенез болезни Гуам связан с формированием многочисленных нейрофибриллярных включений, преобладающих в коре, базальных ганглиях, гиппокампе, миндалевидном теле, спинном мозге, которые приводят к дегенерации соответствующих нейронов. Причиной конформационных нарушений в нейронах считают изменения в гене, кодирующем синтез микротубулярного тау-протеина (Morris et al., 2001; Hasegawa et al., 2007). Наrry Zimmerman (2001) показал, что заболеваемость БАС у коренного населения острова Гуам, расположенного в Тихом океане, составляла около 70 на 100 000 населения и была связана с питанием черепаховым мясом, которое использовали аборигены в пищу. При ограничении этой еды в пищу заболеваемость БАС на Гуам снизилась к концу века с 70 до 7 на 100 000 населения. Этот феномен получил название «комплекс острова Гуам».
Несмотря на то что удалось идентифицировать некоторые генетические факторы риска, фундаментальные причины, лежащие в основе БАС, продолжают оставаться неясными. Недавний пересмотр факторов риска внешней (окружающей) среды, выступающих в качестве этиологии БАС, не позволил прийти к установлению последовательной взаимосвязи между отдельным фактором внешней среды и риском развития болезни. За последнее десятилетие в области изучения этиологии и патогенеза БАС достигнуты определенные успехи.
Одной из красивых гипотез возникновения БАС является нейрофиламентная теория. Ее смысл заключается в том, что моторные нейронные клетки это самые большие нейроны в человеческом организме. Сохранение структуры нейрона и транспортные процессы в нем обеспечиваются цитосклелетом этой клетки. Белки цитоскелета – это в первую очередь нейрофиламенты – микроскопические системы трубочек и каркаса клетки. Мутантная СОД-1 может взаимодействовать с нейрофиламентами, что нарушает аксональный транспорт.
Также одним из малоизученных аспектов патофизиологии БАС является функциональное состояние сегментарного аппарата спинного мозга и внутри- и надсегментарных влияний на периферический мотонейрон. Этот аспект патогенеза БАС представляется актуальным для дальнейшего исследования, поскольку изучение изменений на уровне сегмента спинного мозга может дать возможность обоснования новых методов лечения с целью оптимальной фармакологической коррекции афферентно-эфферентной дисфункции сегментарного уровня. Эта теория получила название гипотезы восходящей и нисходящей нейродегенерации. БАС традиционно рассматривается как заболевание двигательной системы. Однако существуют работы, подтверждающие мультисистемный характер БАС с поражением экстрамоторных отделов центральной нервной системы (ЦНС) (Appel et al., 2008; Ordes et al., 2011; De Vos et al., 2008; Meininger et al., 2000) и экстранейральных систем (Meininger et al., 2004). Когнитивные нарушения, отражающие экстрамоторное поражение мозга, долгое время рассматривались как исключительные клинические варианты случайного развития деменции у 3—5% пациентов с БАС (Beauverd et al., 2012). Но в настоящее время все больше подтверждений находит гипотеза, что БАС и фронтотемпоральная деменция (ФТД) имеют общие компоненты патогенеза или даже являются различными проявлениями одного заболевания. Относительно высокая частота деменций при БАС подчеркивает необходимость разработки методик для краткого нейропсихологического обследования пациентов. Кроме уточнения патогенетических моментов и коррекции ведения пациента с учетом наличия у него когнитивных расстройств и угрозы развития деменции, ментальная сфера пациентов с БАС может стать предметом тщательного обследования, если встает юридический вопрос о дееспособности пациента2. Большинство авторов выступают в поддержку сложносоставных взаимодействий на уровне генетической микросреды, выступающих в качестве предполагаемого причинного фактора дегенерации двигательных нейронов (Shaw, 2005; Cozzolino et al., 2008). Были исследованы гипотетические экзогенные факторы риска БАС, и их список может быть представлен следующими экзогенными факторами риска развития спорадических форм БАС: пищевые (диетические) факторы, электрическая травма, семейный анамнез болезни Альцгеймера или Паркинсона, географическое место жительства, служба в армии, детородный возраст, количество родов (у женщин) и порядок родов, возраст при наступлении менопаузы, потеря ребенка, род занятий, физическая активность, игра в футбол на профессиональном уровне, предыдущие полиомиелитные инфекции, раса и этническая принадлежность, курение, воздействие токсинов (сельскохозяйственные химические вещества, свинец), травмы позвоночника, стаж образования.
Генетические факторы. Примерно у 10% людей с заболеванием БАС имеется по крайней мере еще один член семьи, подвергшийся воздействию данной болезни, и предполагается, что в анамнезе может быть обнаружен семейный след развития БАС. Около 20% больных с аутосомной доминантной формой семейного БАС (СемБАС) и 2% больных с формой спорадического БАС (СпорБАС) обнаруживают мутации гена супероксиддисмутазы меди цинка SOD1 (Rosen et al., 1993). Предполагается, что мутации гена SOD1 вызывают развитие заболевания посредством токсического приобретения функции, а не через механизм утраты антиоксидантной функции фермента SOD1 (Shaw, 2005). К другим генам, вызывающим развитие семейной формы БАС (семБАС), можно отнести: алсин (alsin, БАС2); сенатаксин (senataxin, БАС4 и БАС5); везикуло-ассоциированный мембранный протеин (VAPB, БАС8), ангиогенин (angiogenin) и мутацию, происходящую в подгруппе p150 динактина (dynactin) DCTN1; оптинеурин (optineurin, ALS12). В недавнее время у пациентов с семейной и спорадической формами БАС (семБАС, спорБАС) были обнаружены мутации гена TARDBP, кодирующего TAR ДНК-связывающий протеин 43 (TDP-43), расположенный в хромосоме 1p36.22. Более того, мутации гена fused in sarcoma (FUS) недавно были идентифицированы примерно у 4% больных с семейной формой БАС, негативной к гену SOD1. Некоторые мутации других видов генов, способные увеличивать восприимчивость к БАС, определялись и у спорадических пациентов, в частности, мутации в KSP повторной области гена тяжелого нейрофиламента (NEFH), кодирующего нейрофиламентные тяжелые производные единицы; мутации в генотипе аполипопротеина Е Σ4 (apolipoprotein E Σ4 genotype); мутации в сниженной экспрессии протеина транспортера 2 возбудительной аминокислоты EAAT2. Сообщалось также о связи трех мутаций VEGF гена с повышенным риском развития спорадического БАС (спорадических случаев заболевания), в то время как недавно проведенный многоплановый метаанализ не выявил взаимосвязи между гаплотипами гена VEGF и увеличением риска возникновения БАС у людей. Подобные вариационные изменения отмечались в генах-кандидатах или при выполнении общегеномных исследований ассоциативных связей (Dion et al., 2009).
Курение как патогенетический фактор БАС. Методы доказательной медицины подтвердили связь курения с развитием БАС (Chen Lien al., 2015).
На сегодня не существует общепризнанной гипотезы патогенеза бокового амиотрофического склероза. Согласно современным представлениям, развитие БАС обусловлено взаимодействием наследственных и экзогенных провоцирующих факторов. Множество патологических изменений в нейронах приводит к предположению о многовариантном этиологическом факторе.
Окислительный стресс считается важнейшим фактором в этиопатогенезе БАС. Известно, что окислительный стресс причастен к процессу нейродегенерации при БАС. Также известно, что скопление образцов активных форм кислорода вызывает гибель клеток. Поскольку мутации, происходящие в гене SOD1, могут привести к семейному типу БАС, этот механизм окислительного стресса дегенерации двигательных нейронов при БАС представляет научный интерес. Данная гипотеза поддерживается биохимическими изменениями, отражающими повреждения свободных радикалов и патологический метаболизм свободных радикалов в цереброспинальной жидкости и образцах аутопсий пациентов с БАС (Ferrante et al., 1997; Smith et al., 1998; Tohogi et al., 1999). Кроме того, у образцов фибробластов, культивированных у больных с БАС, выявлялась повышенная чувствительность к окислительным разрушениям по сравнению с участниками контрольных групп (Aguirre et al., 1998).
Предполагается, что перекись водорода может служить аномальным субстратом для конформированной молекулы SOD1. В результате происходит усиление пероксидантных реакций и возрастает производство токсичных гидроксильных радикалов. Существенная роль окислительного стресса в патогенезе БАС подтверждается биохимическими исследованиями, при которых у больных обнаружилась недостаточность ряда систем антиоксидантной защиты, дисфункции митохондрий, дисметаболизм глутатиона, эксайтотоксина глутамата и механизмы глутаматного транспорта. Возможно, окислительное повреждение белковых мишеней (SOD1, нейрофиламентных белков, альфа-синукленина и т.д.) может облегчать и ускорять их совместную агрегацию, формирование цитоплазматических включений, которые служат субстратом для дальнейших патохимических окислительных реакций.
Апоптоз – опосредованная каспазами гибель клеток. Предполагается, что окончательный процесс гибели двигательных нейронов при БАС напоминает апоптозный путь запрограммированной клеточной гибели. Апоптоз представляет собой механизм, посредством которого двигательные нейроны отмирают в моделях мутантных мышей с трансгенным SOD1. В этих моделях были последовательно активированы апоптозные сигналы каспазы-1 и -3. Хроническая активация каспазы-1 происходила в раннем пресимптоматическом периоде, а каспаза-3 была активирована намного позже, будучи финальным эффектором гибели клеток (Pasinelli, 2000). В соответствии с данными сообщениями, внутрицеребровентрикулярная доставка широкого ингибитора каспазы приводила к снижению mRNA уровней каспазы-1 и -3 в тканях спинного мозга мутантных мышей с трансгенным SOD1 (G93A). Кроме того, двигательные нейроны были сохранены у мышей, прошедших трансфузию ингибитором каспазы, в сравнении с мышами, получившими трансфузию носителя-растворителя. Также удалось задержать начало развития заболевания и его прогрессирование (Li et al., 2000). Биохимические маркеры апоптоза определяются в терминальной стадии БАС пациентов и животных моделей. Ключевые элементы пути здорового апоптоза вовлечены в гибель клеток при БАС, в том числе семейство каспаз протеолитических ферментов, Bcl2 семейство онкопротеинов (антиапоптозные и проапоптозные онкогены) и ингибирующее апоптоз семейство протеинов. Что касается дополнительного подтверждения, говорящего в пользу апоптозного механизма гибели нейронов, было замечено, что избыточная экспрессия антиапоптозного протеина Bcl-2 (Kostic et al., 1997) и удаление проапоптозного протеина Bax (Gould et al., 2006) способны удерживать и сохранять двигательную функцию и продлевать выживаемость мутантных мышей с трансгенным SOD1 (G93A). Таким образом, токсичность мутантного SOD1, по всей видимости, опосредуется, по крайней мере частично, каспазами и другими апоптозными факторами.
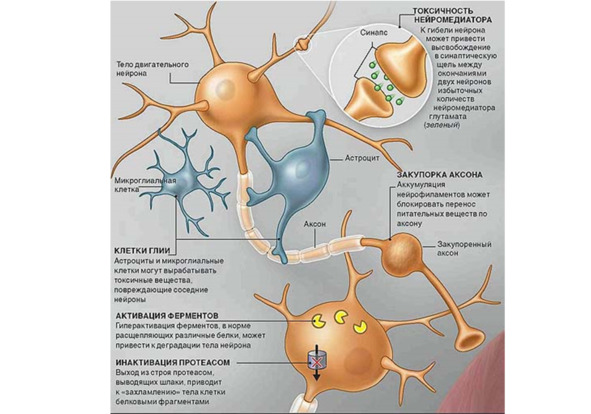
Рис. 1. Установленные негенетические молекулярно-биологические механизмы развития бокового амиотрофического склероза
В интернете мы нашли очень познавательную схему патофизиологических нарушений и молекулярно-биологических механизмов, происходящих в нервной ткани при БАС, и приводим ее для иллюстрации всего вышесказанного (рис. 1).
Представленная схема позволяет очень наглядно увидеть все основные молекулярно-биологические механизмы патогенеза данного заболевания (рис. 2).
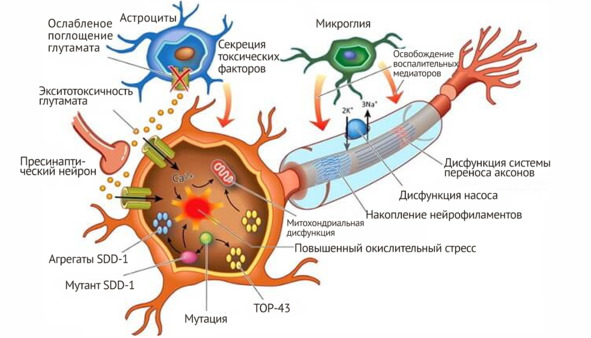
Рис. 2. Схема локальных молекулярных повреждений в двигательном нейроне при боковом амиотрофическом склерозе
Как видно на рисунке 2, локальные молекулярные повреждения в мотонейроне связаны с ослаблением поступления глютамата из астроцитов с формированием экситоксичности глютамата, что приводит к митохондриальной дисфункции в мотонейроне, нарушению обмена внутриклеточного Са2+ и возникновению повышенного окислительного стресса, появлению мутантного фермента СОД-1 и, как следствие, появлению мутаций в других генах ядра нейрона. Эти молекулярно-биологические нарушения приводят к появлению в цитоплазме мотонейрона агрегатов СОД-1, которые усиливают процессы мутагенеза. Освобождение воспалительных медиаторов из клеток микроглии приводит к локальному повреждению структуры аксонов и нарушает их информационную проводимость путем нарушения их цитоскелета.
Все большее число исследований указывают на то, что воспаление в центральной нервной системе, а также повышенная концентрация иммунных клеток, вызывающих воспалительные реакции, обнаруженная в центральной нервной системе пациентов с БАС, являются ключевыми этиопатогенетическими факторами при БАС. Клинические исследования выявили периферическое воспаление в БАС; были обнаружены такие маркеры воспаления, как Т-клетки, цитокины и хемокины. Китайские ученые из Университета Минзу в Китае (Minzu University of China; Пекин) провели систематический обзор и метаанализ 25 исследований, включая исследование 812 пациентов с БАС и 639 человек в контрольной группе. Это было сделано для устранения противоречивых результатов. Сравнивались уровни воспалительных цитокинов пациентов с БАС и пациентов контрольной группы, затем данные клинических исследований объединялись и анализировались. Метаанализ выявил значительную гетерогенность для 8 из 14 цитокинов. Интерлейкин (IL) -2, IL-4, IL-17, фактор роста эндотелия сосудов (VEGF) показали умеренные уровни гетерогенности, тогда как фактор некроза опухоли-α (TNF-α), моноцитарный хемоаттрактантный белок-1 (MCP-1), интерферон гамма (IFNγ), IL-5 показали высокие уровни гетерогенности. Анализ в подгруппах показал, что уровни TNF-α в крови были значительно увеличены у пациентов с БАС по сравнению с пациентами в контрольной группе. Ученые отметили: предыдущие данные показали, что уровни TNF-α, IL-6 и IL-1β в крови повышены у пациентов с болезнью Альцгеймера и болезнью Паркинсона, в то время как рецептор фактора некроза опухоли 1 (TNFR1) увеличен при болезни Паркинсона, что дает представление об общем механизме у различных нейродегенеративных нарушений. Авторы пришли к выводу, что метаанализ впервые использовался для исследования изменений уровней воспалительных цитокинов у пациентов с БАС и показал увеличение уровней TNF-α, TNFR1, IL-1β, IL-6, IL-8 и VEGF в периферической крови у пациентов с БАС по сравнению с пациентами в контрольной группе. В исследовании подчеркивается, что периферические уровни цитокинов могут быть биомаркерами для БАС, что может быть интересным для медицинских работников, стремящихся преодолеть разрыв между постановкой диагноза и появлением симптомов. Исследование было опубликовано 22 августа 2017 г. в журнале Scientific Reports (https://studfiles.net/preview/1818013/page:92/).
Роли дисбаланса трофических факторов в этиопатогенезе БАС в последнее время придается большое значение, и эти инновационные молекулярные исследования оформились в новую теорию трофической недостаточности. Это связано с тем, что нейрональное развитие и выживание нейронов в нервной системе человека зависят от сбалансированной и строго управляемой поддержки, исходящей от трофических факторов. Подобные факторы способны регулировать и управлять такими важными физиологическими процессами, как нейрональная дифференциация, поддержка синапсов, нейрональное выживание, осуществляемое через ингибирование апоптоза, нейрогенез и аксональное прорастание (Korsching, 1993; Boonman, Isacson, 1999; Hou et al., 2008). Кроме того, они создают условия для формирования определенных экологических ниш, пригодных для выживания нейронов (Mudò et al., 2009). Трофическая поддержка имеет первостепенное значение для нейронов спинного мозга и обеспечивается из многочисленных различных клеточных источников, включая астроциты, микроглии, нейроны и эндотелиальные клетки (Ikeda et al., 2001; Béchade et al., 2002; Dugas et al., 2008; Su et al., 2009; Hawryluk et al., 2012). Следовательно, трофическая поддержка рассматривается как перспективная терапевтическая стратегия при нейродегенеративных расстройствах (Kotzbauer, Holtzman, 2006) и способна играть важную роль в клеточных методах лечения, нацеленных на реиннервацию утраченных нейромышечных синапсов (Casella et al., 2010).
БАС (ALS) вызывается выборочной и прогрессирующей потерей спинальных, бульбарных и кортикальных двигательных нейронов, что приводит к необратимому параличу, речевым, глотательным и респираторным нарушениям и, в конечном итоге, гибели индивидуумов при быстром течении болезни. БАС преимущественно спорадичен, и 90% случаев происходят при отсутствии семейной истории заболевания. Тем не менее за последние годы стало очевидно, что многие спорадические случаи сопровождаются изменениями белков, мутации которых были выявлены в семейных случаях, что может по меньшей мере повышать вероятность развития БАС (Deng et al., 2010). Многие из подобных мутаций включают изменения в TAR ДНК-связывающем белке 43 (TDP43) и Fused (FUS) белках в генах саркомы, которые соединяют RNA молекулы (Gordon, 2013; Sreedharan, Brown, 2013), в то время как большинство семейных случаев с паттерном преобладающей аутосомной наследственности вызываются мутациями, происходящими в супероксиддисмутазе 1 (SOD1; Rosen et al., 1993). Трансгенные мыши, выделяющие мутантную форму человеческого SOD1, представляют собой наиболее широко используемую модель для изучения БАС in vivo (Gurney et al., 1994). Трофические факторы рассматриваются в качестве терапевтических мишеней при БАС, когда лечение направлено на восстановление утраченных нейромышечных синапсов и защиту двигательных нейронов от токсичности.