


Полная версия
Каталитический риформинг бензинов. Теория и практика
Как было показано ранее, реакция замыкания кольца является самой медленной стадией дегидроциклизации и контролирует скорость ароматизации парафиновых углеводородов. Скорость реакции дегидроциклизации примерно в 30 раз меньше, чем скорость дегидрирования циклогексана.
Поскольку модуль Тиле изменяется пропорционально квадратному корню из величины скорости реакции, его значение для реакции циклизации будет в первом приближении в 5,5 раза меньше – около 0,06. Это означает, что циклизация может проходить в кинетическом режиме, и ее ускорение возможно за счет уменьшения размера пор и увеличения удельной поверхности носителя.
Снижению диффузионных затруднений способствует также экзотермичность реакции циклизации.
Вместе с тем необходимо учитывать, что быстрые реакции расширения кольца и последующего дегидрирования, являющиеся промежуточными стадиями в ароматизации, будут испытывать все большее диффузионное торможение при уменьшении размера пор, и при определенном размере пор будут снижать общее превращение.
Оптимальная поровая структура должна обеспечивать баланс между реакциями, контролируемыми кинетикой, и реакциями, находящимися под диффузионным контролем.
В работе [46] приведены результаты исследования реакции дегидроциклизации н-гептана на платинорениевых катализаторах в зависимости от величины удельной поверхности и распределения пор носителя. В работе использовался катализатор с размером частиц 0,3–0,75 мм. Целью исследования
было определение поровой структуры катализатора риформинга, обеспечивающей максимальную конверсию н-гептана
в толуол.
Некоторые выводы по материалам исследования представлены ниже. В частности, было показано, что активность катализаторов, имеющих одинаковое содержание металлов, но различную поровую структуру, может отличаться в несколько раз.
1. Отсутствует прямая корреляция между активностью
и величиной удельной поверхности катализатора; при оценке активности должна учитываться поровая структура катализатора.
2. При увеличении температуры процесса различие в активности между катализаторами возрастает, этот эффект противоположен тому, что наблюдается при протекании реакции в кинетическом режиме.
3. Кроме того меняются лидеры: так, катализатор, показавший максимальную активность при 470 С, не является таковым при температуре 510 С, и, наоборот, катализатор-аутсайдер становится лидером при более высокой температуре.
4. Увеличение температуры приводит к росту диффузионных затруднений, и реакция перемещается в поры большего размера. Так, при температуре 470 С реакция протекает в порах с радиусом 3 нм и выше, а при 510 С доступными для реакции становятся поры с радиусом 5 нм и более.
5. При увеличении радиуса пор диффузионные затруднения уменьшаются, процесс сдвигается в сторону кинетического режима, и эффективная константа скорости химического превращения приближается к величине действительной константы скорости, достигаемой в отсутствие диффузионных ограничений; это приводит к более эффективному использованию поверхности поры: удельная активность (моль/с), отнесенная к поверхности поры, увеличивается с ростом радиуса поры.
6. Необходимо иметь в виду, что в исследовании использовался катализатор с размером гранул 0,3–0,75 мм. Очевидно, что на промышленном катализаторе, имеющем средний радиус пор 5 нм, но больший размер частиц (1,6 мм), реакция дегидроциклизации н-гептана протекает во внутридиффузионном режиме.
Глава 8. Механизмы реакций риформинга
Схема Миллса для реакций на бифункциональном катализаторе риформинга.
С5– и С6-дегидроциклизация алканов.
Скелетная изомеризация. Карбениевые и карбониевые ионы
Обнаружение роли кислотного промотора привело к разработке концепции бифункционального катализа, предложенной впервые G. Mills, H. Heineman, T. Milliken, A. Oblad из лаборатории Houdry Process Corporation. Концепция оказалась плодотворной и полезной также для понимания механизмов превращений и разработки эффективных катализаторов для процессов изомеризации н-бутана и н-пентана, гидроизомеризации дизельных фракций, гидрокрекинга различных нефтяных фракций и др.
Основная идея этой концепции – ускорение химических реакций за счет синергизма, обусловленного одновременным присутствием кислотных и металлических центров.
Схема реакций платформинга с участием металлических (М) и кислотных (А) активных центров представлена ниже (рис. 16).
Современная схема дополнена реакциями изомеризации и С6-циклизации на металлических центрах.
В соответствии с предложенной схемой ряд реакций может протекать без участия кислотных центров, и для них необходимы только металлические центры, образованные платиной. К таковым относятся реакции дегидрирования и гидрирования, гидрогенолиза и С6-циклизации.
Изомеризация олефинов, циклоолефинов, гидрокрекинг олефинов и циклоолефинов и С5-циклизация, наоборот, не требуют наличия металлических центров и осуществляются на кислотных центрах катализатора.
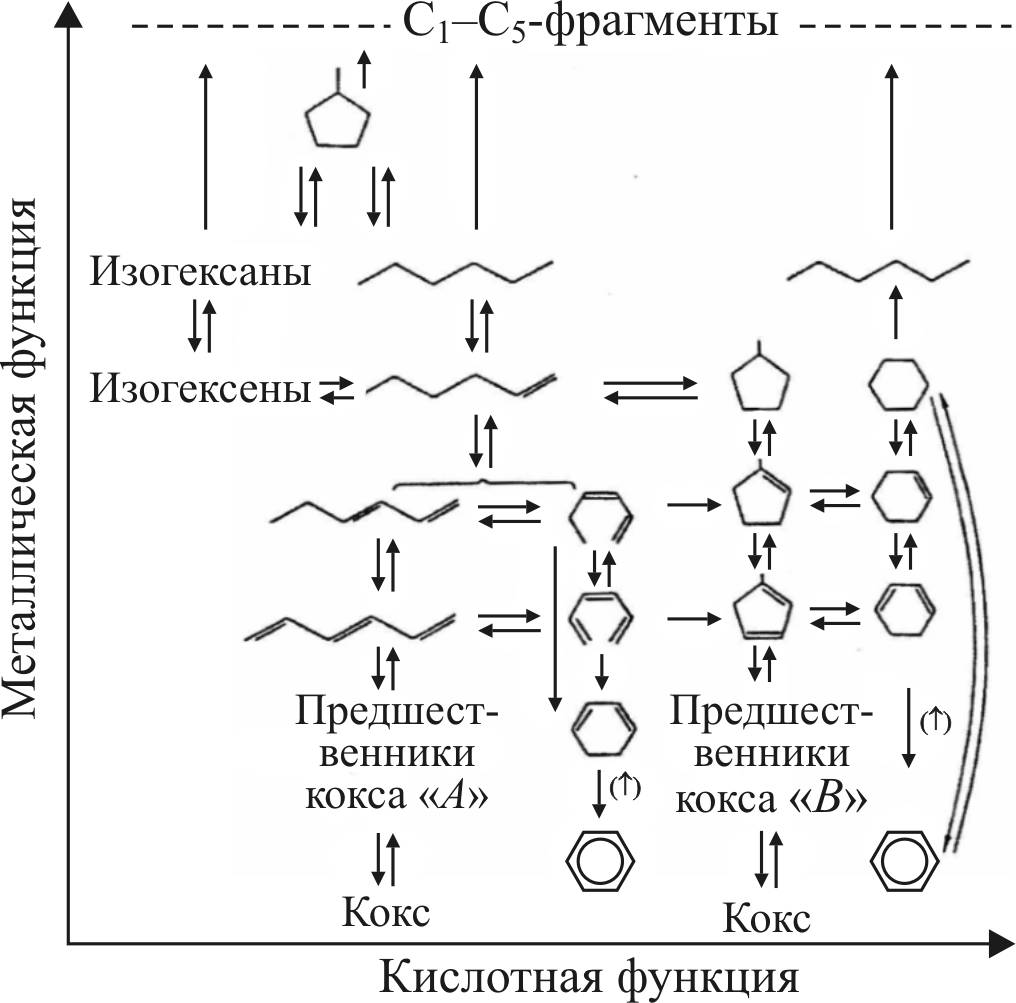
Рис. 16. Схема реакций платформинга по Миллсу
Более сложные реакции проходят с участием активных центров того и другого типа. Например, С5-дегидроциклизация
н-парафинов включает следующие стадии: дегидрирование и образование н-олефина (М), изомеризацию с образованием изоолефина (А), циклизацию изоолефина с образованием алкилциклопентана (А), дегидрирование алкилциклопентана до циклического олефина (М), расширение цикла с образованием 6-членного циклопарафина (А), дегидрирование циклопарафина с получением ароматического углеводорода (М).
Медленными стадиями превращения являются реакции циклизации олефина с образованием 5-членного кольца и последующая реакция расширения цикла. Обе реакции проходят с участием кислотных центров.
Тот факт, что при риформинге н-гексана введение н-бутиламина приводит к снижению концентрации циклогексана при одновременном накоплении метилциклопентана, может свидетельствовать о том, что для осуществления превращений требуются разные кислотные центры [47], в частности, для реакции циклизации кислотные центры Льюиса, представленные ионами Al + 3.
Снижение содержания хлора и, следовательно, бренстедовской кислотности приводит к накоплению молекул циклического олефина (метилциклопентена в случае ароматизации гексана) на поверхности платины и более глубокому дегидрированию с образованием производных циклопентадиена, легко полимеризующегося с образованием предшественников кокса.
Альтернативный маршрут С6-дегидроциклизации, протекающий на катализаторе Pt/C и Pt/KL-цеолит (катализатор RZ-100), в условиях платформинга на кислотном носителе является второстепенным механизмом ароматизации алканов.
Изомеризация н-парафиновых углеводородов включает в себя дегидрирование на металлических центрах с образованием олефинов нормального строения, изомеризацию полученных олефинов на кислотных центрах с образованием олефинов изостроения и гидрирование на металлических центрах до изопарафина. На примере этого превращения становятся понятными преимущества бифункционального катализатора.
Промежуточным соединением в реакции скелетной изомеризации является карбениевый ион, имеющий структуру с трехкоординированным атомом углерода.
В случае олефина образование карбениевого иона происходит достаточно легко при атаке протона, предоставляемого кислотным центром Бренстеда, на π-связь. При этом π-связь разрывается и образуется новая С–Н-связь.
Если же исходным углеводородом является алкан, то
образование карбениевого иона включает атаку протона на
С–Н-связь. При этом вначале образуется промежуточный карбониевый ион, представляющий катион с пятикоординированным атомом углерода. Например, при протонировании 2-метилпентана образуется карбониевый ион (рис. 17, б). Распад этого катиона приводит к образованию карбениевого иона и молекулы Н2 (см. рис. 17, а).
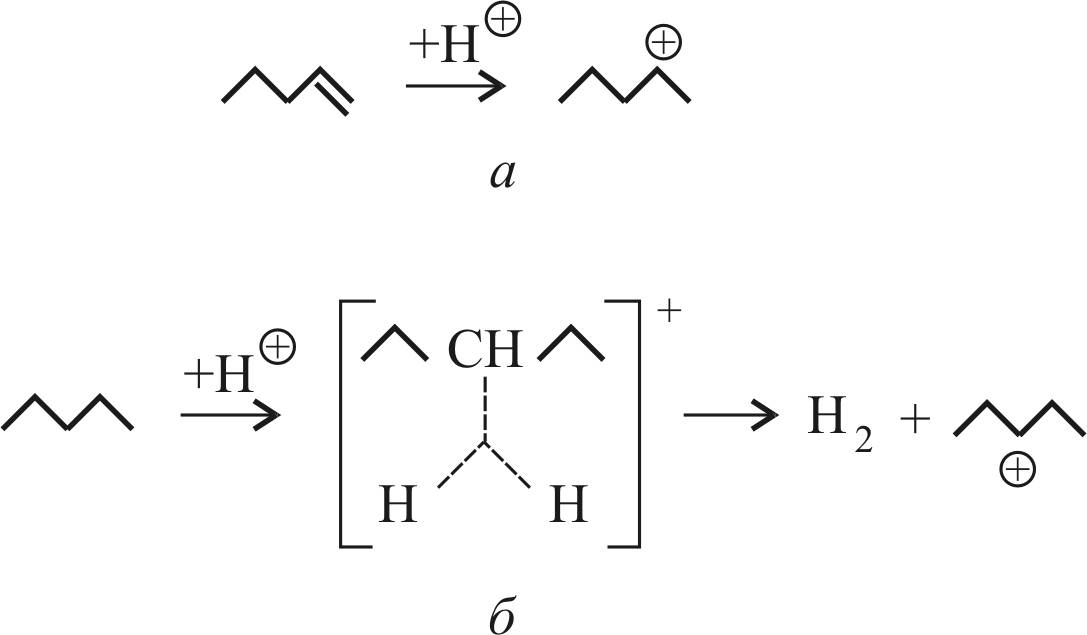
Рис. 17. Схема образования:
а – карбениевого иона; б – карбониевого иона
В этом случае разрывается более прочная С–Н-связь.
Значительная разница в энергиях связи (394–239 = 155 кДж/моль) делает возможным образование карбениевого иона из алкана лишь при использовании твердых суперкислот, например, Н-морденитов с силикатным модулем около 20, в условиях же платформинга подобный механизм не реализуется.
Глава 9. Катализ на
d
-металлах
Природа каталитических свойств d-металлов. Связь с координационной ненасыщенностью поверхностных атомов металлов.
Донорно-акцепторная и дативная связи в металлорганических комплексах. Модель Дьюра – Чата – Дункансона.
Механизм образования связей на примере молекулы СО.
Ослабление связей в молекуле СО как результат образования донорно-акцепторной и дативной связи.
Как образуются d-зоны в металлических катализаторах.
Почему происходит химическая адсорбция.
Активация молекул как результат хемосорбции.
Принцип Сабатье и вулканообразные кривые Баландина.
Почему платина является базовым элементом катализаторов риформинга.
Почему реакции дегидрирования являются быстрыми реакциями, а реакции гидрогенолиза медленными: каталитический эффект платины
Каталитическая активность d-металлов обусловлена координационной ненасыщенностью атомов, образующих поверхностные грани металлических частиц.
Координационное число (КЧ) платины, формирующей гранецентрированную кубическую решетку (ГЦК), равно 12.
КЧ для поверхностных атомов зависят от типа поверхности и составляют 9 для граней (111), 8 для (100) и 7 для (110).
Атомы на ступенях и изломах, ребрах и углах частиц имеют еще меньшие КЧ – от 7 до 5 [54].
Вывод атома из объема металла на поверхность является сильно эндотермическим процессом, связанным с разрывом связей с соседними атомами.
Энергия, необходимая для образования поверхностного атома, прямо пропорциональна энергии когезии металла и координационной ненасыщенности поверхностного атома.
Уменьшение размера частицы также приводит к увеличению поверхностной энергии за счет увеличения доли поверхностных атомов.
Переход системы в более устойчивое состояние с меньшей энергией Гиббса достигается путем коалесценции частиц при повышенных температурах или за счет адсорбции молекул окружающей среды. Из двух видов адсорбции, физической и химической, последняя имеет ключевое значение для гетерогенного катализа, так как связана с активацией молекулы, обусловленной изменениями ее электронной структуры при адсорбции на поверхности твердого тела.
В основе современного понимания механизма химической адсорбции и катализа на d-металлах лежат идеи о координационно-донорной и дативной связях и d-зоне, которые были заимствованы из металлорганической химии и физики твердого тела соответственно.
Дьюар в 1951 году предложил модель образования соли Цейзе и ее палладиевого аналога, комплекса Караша, представляющих собой комплексы этилена и металла (рис. 18) [37].
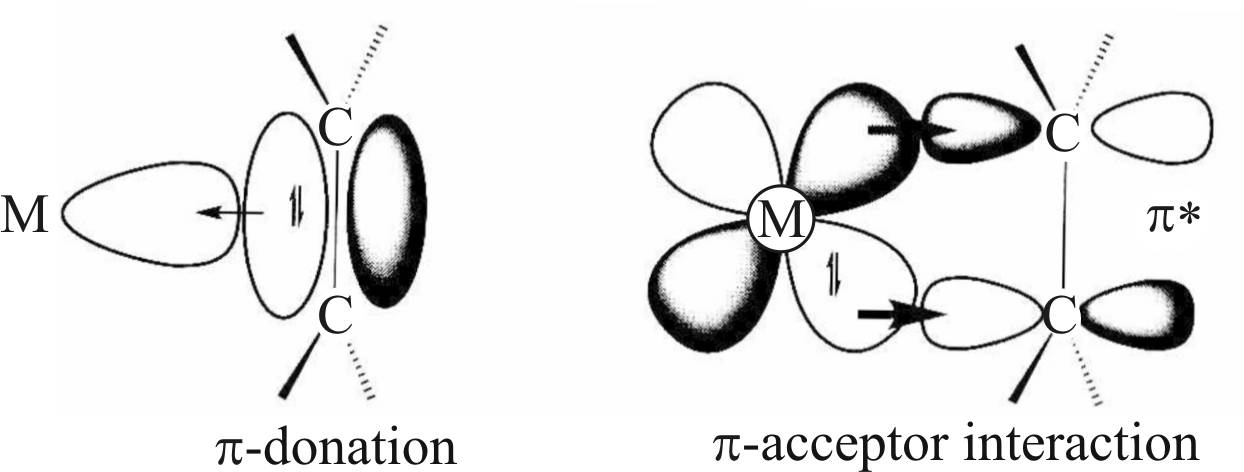
Рис. 18. Структура комплексов Pt(Pd) c этиленом
В соответствии с этой моделью, в доработанном виде носящей название модели Дьюара – Чата – Дункансона, в образовании комплекса принимают участие два типа связей: донорно-акцепторная связь, образуемая за счет передачи электронной плотности π-связи молекулы этилена на вакантную d-орбиталь атома платины, и дативная связь, которая возникает за счет перекрытия заполненной d-орбитали атома металла с разрыхляющей орбиталью молекулы этилена. Заметим, что обе связи являются примером донорно-акцепторного взаимодействия, так что выделение дативной связи сделано для удобства, это указание на то, что донором в этом случае является металл.
Атомными орбиталями, удовлетворяющими этому требованию, являются dz2– и dxz-орбитали переходного металла.
Ниже представлены схемы образования донорно-акцепторной и дативной связей d-металла и молекулы этилена (рис. 19). Донорно-акцепторная связь образуется при перекрывании π-МО этилена с dz2-AO металла. В образовании дативной связи участвуют разрыхляющая π*-МО этилена и dxy-АО металла.
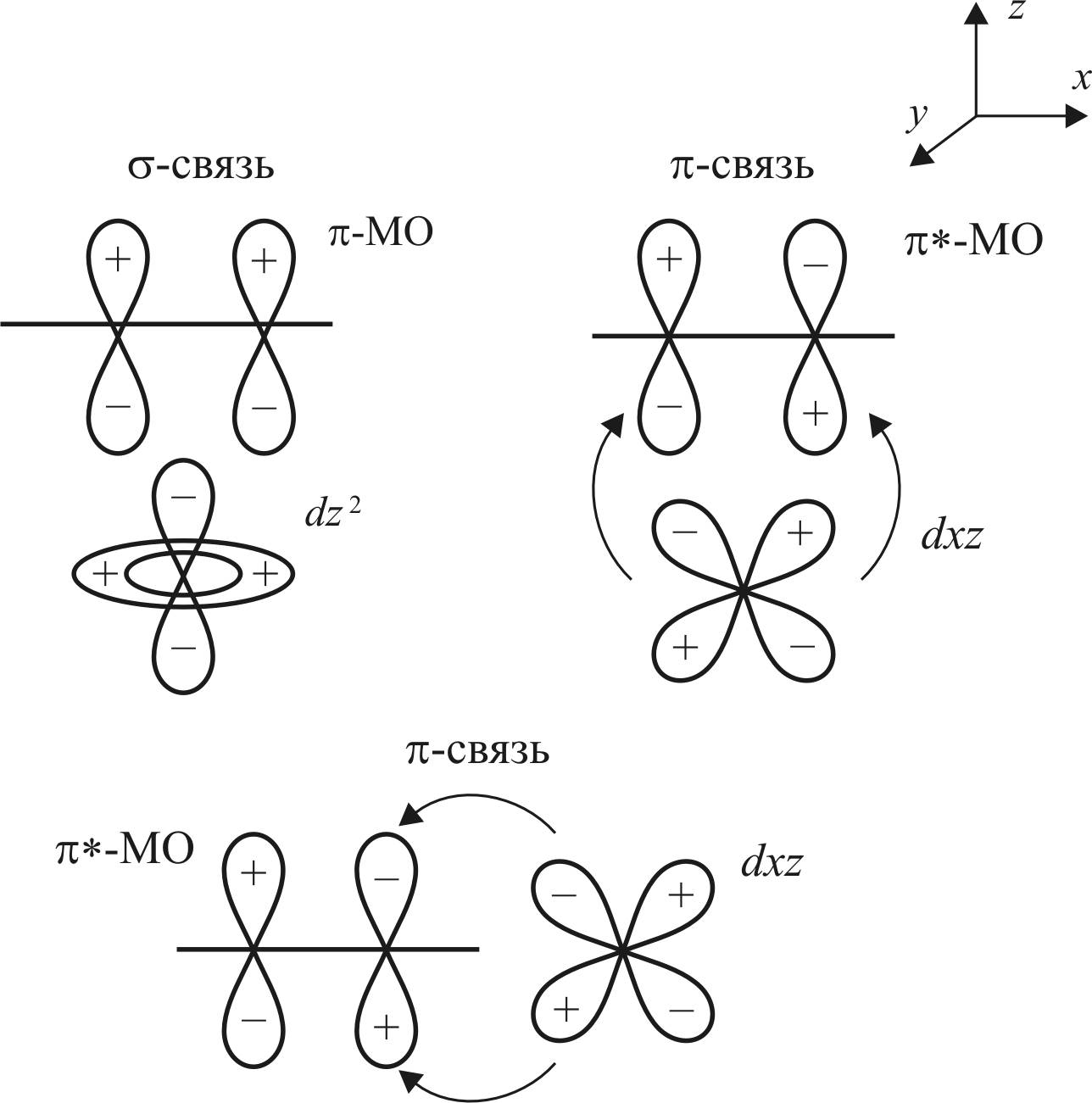
Рис. 19. Схема образования – и π-связей:
стрелками показаны направления смещения электронной плотности
Образование донорно-акцепторной связи осуществляется по σ-типу, а дативной связи – по π-типу.
Для образования дативной связи возможны два варианта перекрывания орбиталей.
Из-за небольших стерических затруднений, возникающих при боковом перекрывании, в рассмотренных комплексах реализуется схема с расположением иона металла над или под плоскостью, в которой находятся sp2-орбитали молекулы этилена.
При отсутствии таких ограничений может реализовываться схема с боковым перекрыванием, например, при образовании связи с молекулой СО, где такому перекрыванию способствует также несимметричное распределение электронной плотности в лепестках разрыхляющей орбитали, связанное с поляризацией связи.
Прочность донорно-акцепторной и дативной связи увеличивается с уменьшением различия в энергии донорной и акцепторной орбиталей в соответствии с величиной энергии стабилизации 106; 107:
ΔЕстаб ~ S2/Δε,
где S – интеграл перекрывания; Δε – разница в энергии исходных орбиталей.
При переносе электронной плотности с π-орбитали этилена происходит накопление положительного заряда в молекуле, что ограничивает перенос электронов, в то же время обратный перенос с занятых орбиталей металла нейтрализует этот заряд, и позволяет продолжить формирование более прочной донорно-акцепторной связи.
В свою очередь передача электронной плотности с молекулы на металл увеличивает донорные свойства металла.
В итоге имеет место синергизм, который приводит к образованию более прочной связи металла и молекулы и более значительному ослаблению связи в молекуле.
Для комплексов Цейзе и Караша связывание является слабым из-за пониженного дативного потенциала положительно заряженных ионов платины и палладия.
Результатом рассмотренных взаимодействий является уменьшение порядка и прочности углерод-углеродной связи в молекуле, что коррелирует с увеличением длины связи и со смещением пиков инфракрасного поглощения в длинноволновую область спектра.
Так, длина связи С–С увеличивается со 133,7 пм в свободной молекуле этилена до 137,0 пм в комплексе платины и этилена и до 148,0 пм в комплексе этилена и никеля Ni(CO)4 [37].
Квантово-механический анализ взаимодействия на примере связывания молекулы СО и переходных металлов первой серии (3d-металлы) представлен в [106].
Электронная структура молекулы СО представлена на рис. 20.
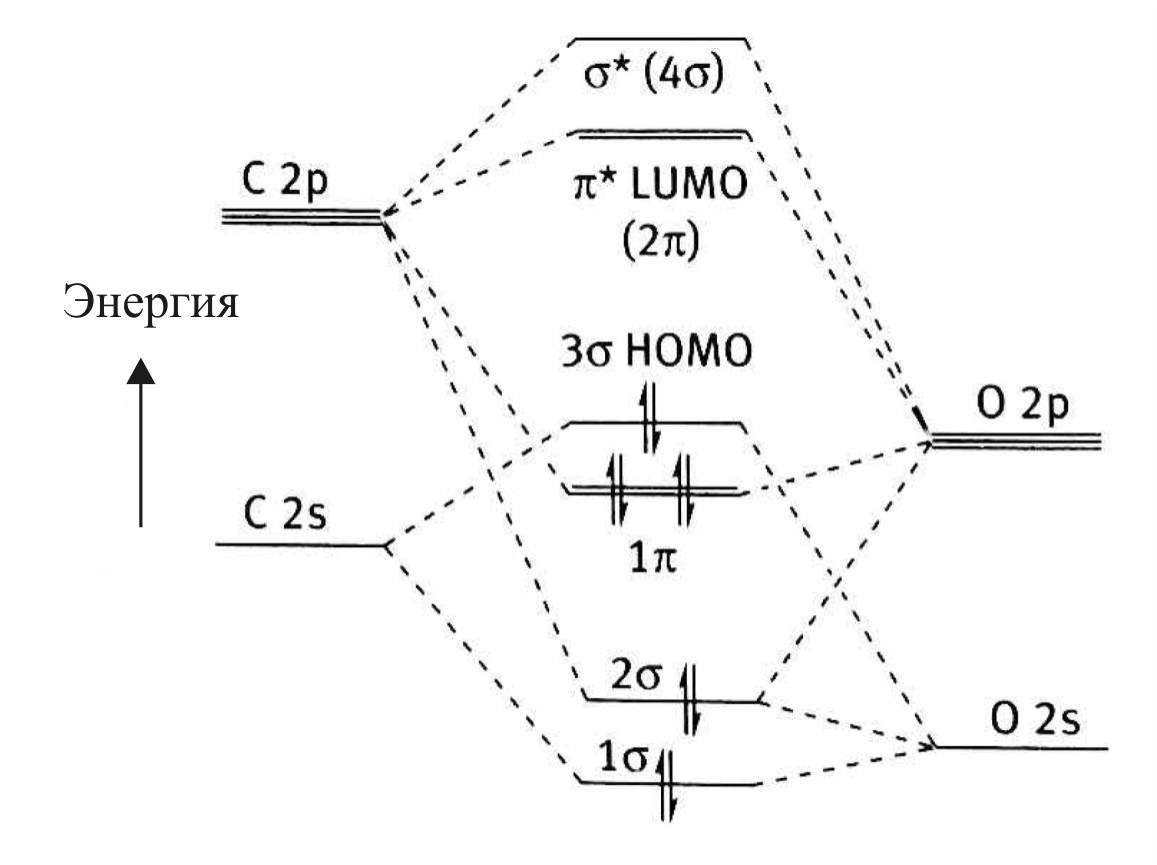
Рис. 20. Электронная структура молекулы СО:
HOMO – high occupated molecular orbital;
LUMO – low unoccupated molecular orbital
В образовании связей в молекуле принимают участие
1σ-орбиталь и две 1π-орбитали.
Молекулярные орбитали 2σ и 3σ не вносят вклада в связывание и являются фактически не поделенными электронными парами кислорода и углерода соответственно.
На самом деле орбиталь 3σ является слегка разрыхляющей орбиталью для молекулы. Эта орбиталь участвует в донорно-акцепторном связывании с d-металлом в качестве донора электронов.
Особенностью электронного строения молекулы СО является несимметричное распределение электронной плотности между лепестками π- и π*-МО. Несимметричность обусловлена различиями в электроотрицательности и в уровнях энергии атомов углерода и кислорода, образующих молекулу.
π-МО является связующей в молекуле, и по энергии она ближе к энергии p-AO кислорода, в связи с чем электронная плотность смещена в лепесток у атома кислорода.
π*-орбиталь как разрыхляющая орбиталь молекулы ближе по энергии к p-орбитали атома углерода, что обусловливает концентрацию электронной плотности орбитали в лепестке у атома углерода (рис. 21).
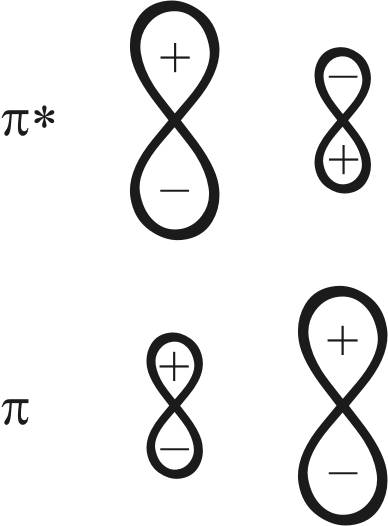
Рис. 21. Электронные схемы π*-, π-орбитали
В связи с тем что σ-связывание с металлом происходит через атом углерода, подобное несимметричное распределение π-электронной плотности более благоприятно для связывания dxz-орбитали металла с π*-орбиталью (рис. 22).
Расчетное отношение интегралов перекрывания для π- и π*-орбиталей составляет 1,78 в пользу π*-орбитали.
Поскольку энергия стабилизации при образовании связи пропорциональна квадрату интеграла перекрывания, то выигрыш в энергии при образовании связи с π*-орбиталью больше в 3,2 раза. Такой выигрыш благоприятен для проявления π-акцепторного характера π*-МО. Вместе с тем необходимо учитывать различие в энергиях орбиталей Δε.
Значения Δε представлены в табл. 5.
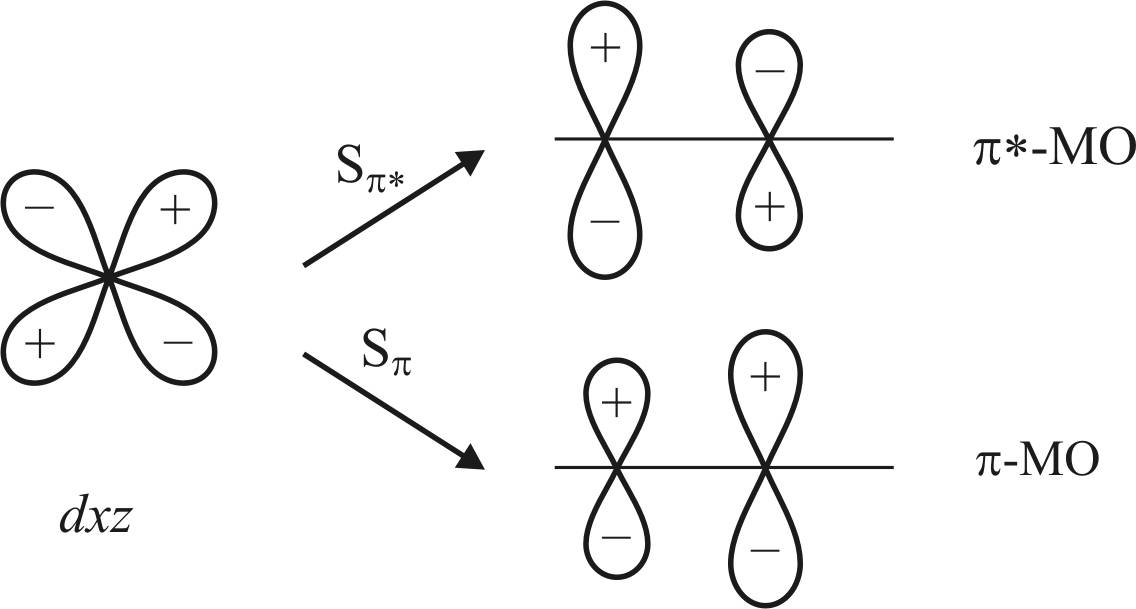
Рис. 22. Схема dxz-орбиталей атома металла
с π*-МО и π-МО молекулы СО
Таблица 5
Энергетические уровни d-металлов
и орбиталей молекулы СО [106]
Энергия
Sc
Ti
V
Cr
Mn
Fе
Co
Ni
Сu
d
–8,5
–10,8
–11,0
–11,2
–11,7
–12,6
–13,2
–13,5
–14,0
7,2
4,9
4,7
4,5
4,0
3,1
2,5
2,2
1,7
*
–0,6
1,7
1,9
2,1
2,6
3,5
4,1
4,4
4,9
П р и м е ч а н и е : εd – энергия d-электронов в атоме металла.
Напомним, что нулевому энергетическому уровню соответствует состояние электрона, находящегося на таком удалении от ядра, когда можно пренебречь электростатическим взаимодействием электрона и ядра атома.
При приближении электрона к ядру его потенциальная энергия падает, поэтому чем больше по величине отрицательное значение εd, тем ниже энергия электрона.
Для металлов в левой части периода d–π*-взаимодействие оказывается сильнее d–π-взаимодействия, и для этих металлов молекула СО выступает как акцептор электронов.
Для металлов в правой части периода разница в энергии благоприятствует проявлению донорного характера π-связи, однако больший интеграл перекрывания с π*-связью приводит все же к тому, что молекула остается π-акцептором электронов.
При переходе к металлам второго и третьего периодов происходит подъем энергетического уровня d-электронов, что приводит к сближению π*-МО и d-электронов и увеличению
π-акцепторного характера молекулы СО (табл. 6).
Таблица 6
Энергетические уровни d-орбиталей первого, второго
и третьего переходного периодов [106]
Первый период
Sc
Ti
V
Cr
Mn
Fе
Co
Ni
Сu
Zn
3d
–7,92
–9,22
–10,11
–10,74
–11,14
–11,65
–12,12
–12,92
–13,46
–17,29
4d
–6,60
–7,11
–7,32
–7,45
–7,83
–7,90
–8,09
–8,22
–8,42
–9,39
Второй период
Y
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Аg
Cd
4d
–6,48
–8,30
–8,85
–9,14
–9,25
–9,31
–9,45
–9,58
–12,77
–17,85
3d
–6,70
–7,31
–7,22
–7,24
–7,21
–7,12
–7,28
–7,43
–7,57
–8,99
Третий период
Lu
Hf
Та
W
Re
Os
Ir
Pt
Au
Hg
5d
–5,28
–6,13
–7,58
–8,76
–9,70
–10,00
–10,21
–10,37
–11,85
–15,58
6d
–7,04
–7,52
–8,45
–8,51
–8,76
–8,81
–8,83
–8,75
–9,22
–10,43
П р и м е ч а н и е : приведенные значения энергий рассчитаны на базе спектроскопических данных.
Формально образование связей с атомом металла может быть представлено уравнением с использованием структур Льюиса:
Структура образующегося комплекса является резонансом двух указанных структур. Обе структуры имеют меньшую энергию, чем исходные реагенты: первая за счет образования новой связи, а вторая стабилизирована дополнительно, потому что в ней отсутствует разделение зарядов.
Порядок связи в результирующей структуре находится между 3 и 2, то есть меньше, чем в исходной молекуле, что также свидетельствует, что образование связей с атомом металла приводит к ослаблению связи в молекуле СО. При переходе от концевых к мостиковым группам связь в молекуле СО ослабевает еще больше, что проявляется в смещении ИК-поглощения в длинноволновую часть спектра (рис. 23) [37].
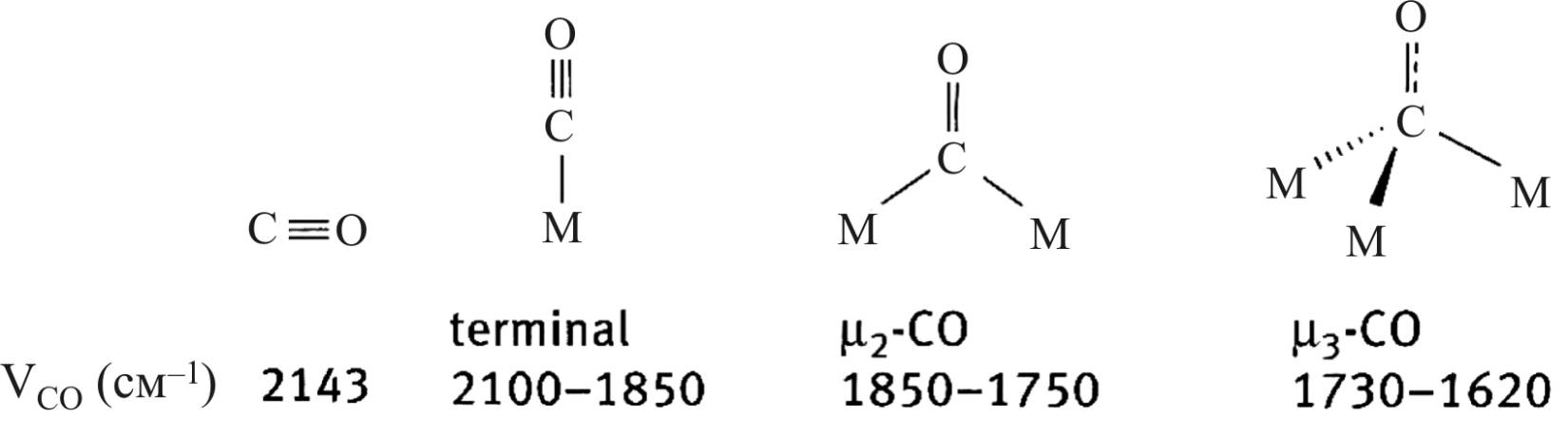
Рис. 23. Структура адсорбционных комплексов
молекулы СО и атомов металлов
Этот пример из металлорганической химии имеет прямую аналогию в гетерогенном катализе и позволяет понять, почему увеличение размера активного ансамбля приводит к более легкому протеканию структурно-чувствительных реакций.
Сказанное выше не означает, что молекула СО не может быть донором электронов для атома металла.
Наличие в молекуле 3σ-орбитали, имеющей характер неподеленной электронной пары, причем обладающей небольшим разрыхляющим эффектом, позволяет молекуле СО выступать в качестве σ-донора по отношению к элементам, имеющим незаполненные d-орбитали.
Необходимо учитывать, что преобладающий вклад дативного взаимодействия обусловлен поляризацией связи С–О, являющейся причиной высокого значения интеграла перекрывания d–π*.
Для молекул с меньшей поляризацией связей этот эффект будет меньше или вообще отсутствовать, например, дипольный момент связи С–С равен нулю. Вместе с тем связь С–Н существенно поляризована: дипольный момент связи составляет 0,3 дебая (Д), что почти в три раза больше, чем дипольный момент молекулы СО (0,11 Д).
Такие различия в поляризации, как будет показано далее, вполне могут быть причиной различных скоростей реакций дегидрирования и гидрогенолиза углеводородов, протекающих на катализаторе платформинга.
После рассмотрения координационной связи в металлорганических соединениях перейдем к анализу взаимодействия молекул с поверхностью металла. Это взаимодействие лежит в основе химической адсорбции и определяет каталитическое действие переходных металлов.
Основным отличием взаимодействия с поверхностью является то, что молекула образует связи не с орбиталями отдельного свободного атома металла, а с энергетическими зонами, возникающими при перекрывании орбиталей отдельных атомов. Это создает дополнительные возможности для образования химической связи, как мы увидим далее.
Формирование энергетических зон может быть описано двумя различными способами.
В приближении свободных электронов (ПСЭ) или модели электронного газа, используемых в физике твердого тела, зоны формируются аналогично тому, как происходит квантование энергетических уровней электрона, помещенного в прямоугольную потенциальную яму [11; 25].
Наличие периодической решетки, образованной атомами, приводит к расщеплению континуума энергий электронов на серию зон Бриллюэна.
В приближении сильной связи (ПСС) зоны формируются при перекрывании орбиталей атомов аналогично тому, как это происходит при образовании молекулы.
На рис. 24 представлено образование s-зоны для n атомов лития. Аналогичным образом формируются s-, p– и d-зоны для d-элементов.
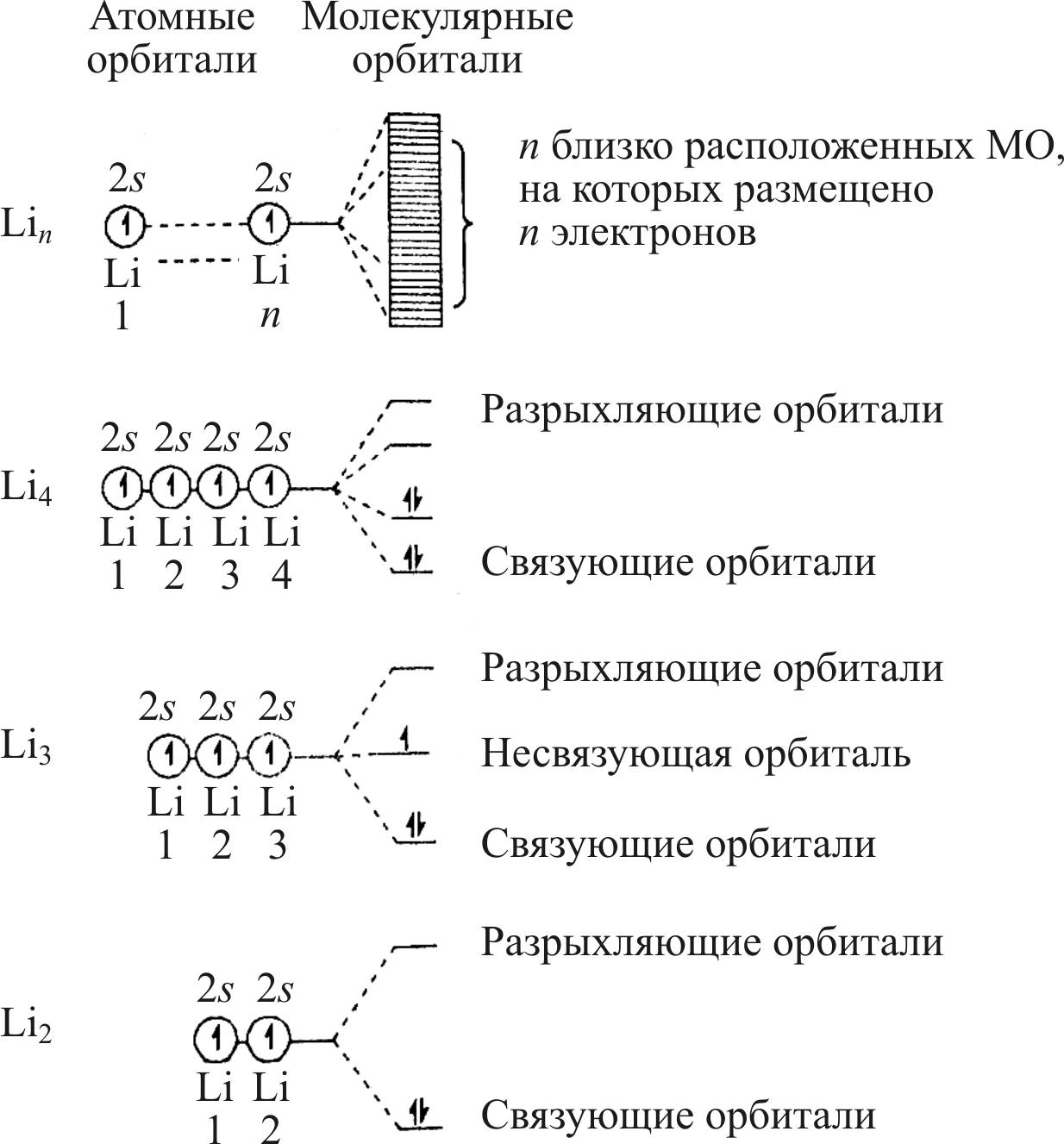
Рис. 24. Схема образования s-зоны для n атомов лития