


Полная версия
Диффузные болезни соединительной ткани
Таким образом, поражение миокарда при ДМ (ПМ) может протекать в виде выраженного кардита, но чаще развиваются субклинические варианты, что диктует необходимость использования современных методов функционального исследования сердца для выявления и уточнения характера его поражения.
Новые неинвазивные методы исследования сердца позволили подтвердить частоту и различный характер его поражения при ДМ (ПМ). Так, при использовании данных эхокардиографии (ЭхоКГ), суточного мониторирования, перфузионной сцинтиграфии с 201Т1 и исследовании центральной гемодинамики были выявлены изменения со стороны сердца у всех обследованных больных. Одновременно у них отмечался и высокий уровень МВ фракции креатинкиназы в крови.
Как показали наблюдения Н. Г. Гусевой (2004), прижизненная биопсия и патологоанатомические исследования в миокарде больных ДМ (ПМ) обнаруживают изменения, в значительной степени сходные с таковыми в скелетных мышцах – мононуклеарную инфильтрацию, иногда некроз и атрофию мышечных волокон. Генез этих изменений при ДМ (ПМ) объясняется наличием миокардита, но, возможно, обусловлен и ишемическими изменениями в связи с поражением мелких сосудов коронарного русла. Для обозначения этой патологии иногда используется термин «полимиозитная кардиопатия».
Описаны случаи дилатационной кардиомиопатии. При высокой клинико-лабораторной активности может наблюдаться развитие констриктивного перикардита. Поражение легких у больных ДМ обусловлено рядом факторов, среди которых выделяют мышечный синдром (гиповентиляция), присутствие инфекционных агентов, аспирацию при нарушении глотания, развитие интерстициальной пневмонии и фиброзирующего альвеолита.
Мышечная слабость, распространяющаяся на дыхательные мышцы, включая диафрагму, может быть причиной снижения вентиляционной функции легких. Клинически отмечается частое и поверхностное дыхание, инспираторная одышка, развивается гипостатическая пневмония. Дисфагия с аспирацией жидкости и пищи в легкие обусловливает развитие аспирационной пневмонии. Поражение легких выявляется у 5 – 46 % больных ДМ (ПМ), главным образом в виде интерстициальной пневмонии, фиброзирующего альвеолита и фиброза. Диагностируется на основании клинико-рентгенологических, функциональных и морфологических (биопсия) данных.
Одышка и кашель, хрипы и крепитация наблюдаются при выраженном поражении легких. Легочные функциональные тесты указывают на преимущественно рестриктивный тип нарушений со снижением общей и жизненной емкости легких; гипоксемия характеризуется умеренным снижением диффузионной способности легких.
Патология легких при ДМ (ПМ) имеет сложный генез и варьирует в широких пределах. Выделяют определенные субтипы интерстициального поражения легких, которые следует учитывать при диагностике и лечении ДМ (ПМ):
I. Острый или подострый тип, который характеризуется тяжелой быстропрогрессирующей одышкой и нарастающей гипоксемией уже в первые месяцы заболевания, аналогично синдрому Хаммена – Рича.
II. Хронический тип с медленнопрогрессирующей одышкой.
III. Асимптомный тип, который протекает субклинически, выявляется при рентгенологическом и функциональном исследовании легких.
Первый тип интерстициального поражения легких имеет наихудший прогноз и требует ранней активной терапии (глюкокортикостероидами (ГКС), цитостатиками и др.).
Полимиозит может предшествовать легочной патологии, развиться после поражения легких или возникать одновременно с легочными проявлениями. Выделяется также и «амиопатический» ДМ, который может ассоциироваться с тяжелым быстропрогрессирующим поражением легких.
Легочный фиброз, обусловленный интерстициальным поражением ткани легких, легочным васкулитом и развитием септально-альвеолярного склероза, отмечается у 5 – 10 % больных. Он характеризуется нарастающей инспираторной одышкой, сухим кашлем, крепитирующими хрипами в нижних отделах легких, нарастающей дыхательной недостаточностью.
Необходимо иметь в виду возможность развития опухолевого, чаще метастатического, процесса в легких.
Поражения желудочно-кишечного тракта (ЖКТ) отмечаются нередко и проявляются нарастающей дисфагией, отсутствием аппетита, иногда – болью в животе и гастроэнтероколитом.
Ведущей симптоматикой поражения желудочно-кишечного тракта при ДМ (ПМ) является дисфагия. Дисфагия развивается вследствие снижения контрактильной силы фарингеальных мышц и мышц верхнего отдела пищевода, нарушения перистальтики, слабости мышц мягкого нёба и языка. Это обусловливает поперхивание, нарушение глотания твердой и жидкой пищи, которая может выливаться через нос. Фарингеально-пищеводная дисфагия – важный дифференциально-диагностический признак ДМ (ПМ). Голос приобретает носовой оттенок. Дисфония нередко сочетается с дисфагией и у тяжелобольных иногда переходит в афонию.
В отличие от ССД при ДМ (ПМ) поражаются верхние отделы пищевода и глоточное кольцо, поэтому клиническая и рентгенологическая картины различны. При склеродермии жидкая пища проходит хорошо, не выливается через нос, однако рентгенологические признаки поражения и осложнения склеродермического эзофагита выражены сильнее, чем у больных с ДМ (ПМ).
Тяжелая прогрессирующая дисфагия, когда твердая пища срыгивается, а жидкая выливается через нос, из-за возможности аспирации представляет непосредственную угрозу жизни больного и является прямым показанием к срочной терапии максимальными дозами кортикостероидов.
При вовлечении в процесс пищеводного сфинктера возможно развитие рефлюкс-эзофагита.
Описаны отдельные случаи ДМ (ПМ) с желудочно-кишечными кровотечениями, перфорацией желудка, в основе которых лежат васкулит и некрозы, возникающие в пищеварительном тракте.
Умеренное увеличение печени с изменением функциональных проб наблюдается приблизительно у 1/3 больных; реже выявляется гепатолиенальный и железисто-селезеночный синдромы.
Поражения почек при ДМ (ПМ) встречаются относительно редко. При остром течении тяжелая персистирующая миоглобинурия может привести к развитию почечной недостаточности. Среди больных ДМ (ПМ) у 41,5 % отмечается транзиторная протеинурия с микрогематурией и цилиндрурией (Мазуров В. И., Беляева И. Б., 2005). При остром течении ДМ (ПМ) у отдельных больных могут наблюдаться развитие гломерулонефрита, сосудистая патология почек с фибриноидными изменениями артериол и тромбозом.
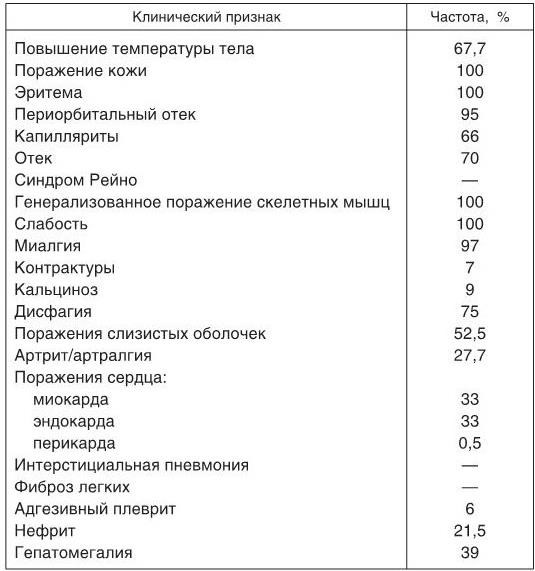
Таблица 1
Частота основных клинических проявлений ДМ (ПМ)
Эндокринные нарушения проявляются изменениями функциональной активности половых желез, гипофизарно-надпочечниковой системы, которые могут быть связаны как с тяжестью заболевания и васкулитом, так и с проводимой стероидной терапией.
Поражение нервной системы встречается редко и характеризуется развитием псевдоневрологической симптоматики, хотя у отдельных больных возможно развитие слабо выраженного полиневрита и даже поражений центральной нервной системы (ЦНС) за счет васкулита. Наиболее часто отмечаются вегетативные расстройства. Изредка наблюдаются нарушения психики, эмоциональная неустойчивость больных, которые могут быть связаны с приемом высоких доз ГКС. Частота основных клинических проявлений ДМ (ПМ) по данным Н. Г. Гусевой (2004) представлена в табл. 1.
1.4. КЛИНИКО-ИММУНОЛОГИЧЕСКИЕ ПОДТИПЫ ДЕРМАТОМИОЗИТА
Кроме классических вариантов ДМ и ПМ можно выделить несколько клинико-иммунологических подтипов, развитие которых ассоциируется с синтезом различных типов антител. Эти подтипы различаются не только по спектру клинических проявлений, но и по иммуногенетическим маркерам, прогнозу, ответу на применение ГКС.
Синтез антител к аминоацил-тРНК-синтетазам ассоциируется с развитием так называемого антисинтетазного синдрома, для которого характерны следующие основные признаки: острое начало миозита, интерстициальное поражение легких, лихорадка, симметричный артрит, синдром Рейно, «рука механика», неполный ответ на применение ГКС с частым обострением при снижении дозы препарата, начало заболевания преимущественно в весеннее время. Характерным проявлением антисинтетазного синдрома является интерстициальное поражение легких, которое выявляется у 50 – 70 % больных с наличием антител Jо-1. Развитие артрита чаще наблюдается у больных с наличием антител Jо-1 (57 – 100 %), чем у больных с другими формами миозита. Артрит, как правило, неэрозивный, характеризуется наиболее частым вовлечением в процесс мелких суставов кистей и лучезапястных суставов. Синдром Рейно при антисинтетазном синдроме наблюдается в 60 % случаев.
При воспалительных миопатиях, сопровождающихся синтезом антител SRP (неантисинтетазные цитоплазматические антитела), чаще выявляются клинические признаки ПМ. Отмечаются следующие клинические особенности: более частое поражение мужчин, чем женщин (6: 1), острое начало и тяжелое течение миозита, высокая частота поражения сердца, плохой ответ на применение ГКС.
Антитела РМ-1, относящиеся к группе антиядерных антител, наиболее часто выявляются при перекрестном синдроме ПМ – ССД. Реже эти антитела выявляются при ювенильном ДМ.
Особенностями миозита «с включениями» являются развитие слабости и атрофии не только проксимальных, но и дистальных групп мышц, умеренное или минимальное увеличение мышечных ферментов, редкая ассоциация с ДБСТ и злокачественными новообразованиями, резистентность к лечению ГКС.
Клинические особенности ПМ, ДМ и миозита с включениями представлены в табл. 2.
При миозите, ассоциирующемся с ДБСТ, в клинической картине превалирует мышечная слабость. Среди заболеваний соединительной ткани, сочетающихся с ДМ (ПМ), на первом месте стоит ССД, затем ревматоидный артрит (РА) и СКВ, реже наблюдаются сочетания с синдромом Шегрена, узелковым полиартериитом (УП), а также с саркоидозом и др.
Таблица 2
Основные клинические симптомы дерматомиозита, полимиозита и миозита с включениями (Насонов Е. Л., 2005)

Существуют три варианта сочетания ДМ (ПМ) с другими заболеваниями соединительной ткани. Первый, когда к картине ДМ (ПМ) присоединяется отдельный(ые) признак(и) другого заболевания, например СКВ или ССД. При втором варианте имеются одновременно признаки ДМ (ПМ) и другого ревматического заболевания, образуя смешанное заболевание соединительной ткани, или overlap-синдром. Третий – развитие ДМ (ПМ) на фоне других диффузных болезней соединительной ткани, например ССД или СКВ, когда ДМ (ПМ) является синдромом основного заболевания. В каждом из таких наблюдений необходимо исключать симптоматику, сходную с другими ревматическими заболеваниями. Так, кожные изменения, особенно при выраженной эритеме и трофических нарушениях, близки наблюдающимся при хронической красной волчанке, а при наличии отека и маскообразности являются склеродермоподобными. Мышечный синдром (без кожных изменений) нередко трактуется как ревматическая полимиалгия, первичная фибромиалгия, а в последнее время и как диффузный фасциит. Общая картина заболевания, наблюдение и использование диагностических критериев позволяют правильно диагностировать ДМ (ПМ).
В основе наблюдающегося полиморфизма ДМ (ПМ), особенно его сочетаний с диффузными заболеваниями соединительной ткани, лежат иммуногенетические особенности, которые проявляются «неравновесным сцеплением» и комбинацией не только определенных генов, ответственных за заболевание, но и связанных с ними клинических симптомокомплексов, формирующих overlap-синдром. Этим можно объяснить и наличие у одного больного признаков трех и даже четырех ревматических заболеваний, включая ДМ (ПМ).
Миозит, ассоциирующийся с опухолями, составляет 20 % всех случаев идиопатической воспалительной миопатии (ИВМ). Частота сочетания злокачественных опухолей с ДМ (ПМ) колеблется от 6,7 до 34 %, что в значительной мере зависит от возраста наблюдающихся больных. Хотя при ряде системных заболеваний соединительной ткани отмечаются определенные ассоциации с неоплазмой (при ССД, синдроме Шегрена и др.), ДМ (ПМ) представляет наиболее яркий и характерный пример паранеопластического процесса при ревматических болезнях. Частота возникновения опухолей при ДМ в 5 – 11 раз выше, чем в общей популяции. Однако у детей сочетание ДМ (ПМ) с опухолями наблюдается редко. В последующие возрастные периоды ассоциация ДМ с неоплазмой возрастает и достигает 40 – 50 % у больных старше 40 лет. Таким образом, развитие ДМ (ПМ) в возрасте после 40 лет может рассматриваться как маркер опухолевого процесса.
Сочетание ДМ (ПМ) с неоплазмой встречается чаще у мужчин, чем у женщин, хотя среди наблюдавшихся Н. Г. Гусевой это преобладание не столь значительно. При сочетании с опухолевым процессом превалирует картина ДМ, а не ПМ, что может быть обусловлено генерализованным характером паранеопластических реакций. При этом следует отметить, что ПМ чаще отмечается у женщин.
Как один из механизмов развития опухолевого ДМ (ПМ) обсуждается возможность перекрестных реакций между неопластическим и мышечным антигенами, активация латентной вирусной инфекции с последующим включением иммунных механизмов и развитием ДМ (ПМ), воздействие вирусного или иных факторов на иммунный аппарат с экспрессией одновременно ДМ и опухолевого роста. Антигенное воздействие опухоли и включение иммунных механизмов развития паранеопластического ДМ (ПМ), очевидно, возможны на самых ранних этапах бластогенеза, когда тщательное общеклиническое исследование не обнаруживает неоплазмы, что не является доказательством ее отсутствия.
Чаще злокачественную опухоль выявляют во время обследования больного с мышечной слабостью или уже установленным диагнозом ДМ (ПМ), при этом мышечная слабость как проявление ДМ (ПМ) может предшествовать обнаружению неоплазмы на 1 – 2 года и раньше. Реже опухоль диагностируют до появления признаков ДМ (ПМ).
Характер локализации неоплазм при ДМ (ПМ) отражает общие возрастные и популяционные закономерности, за исключением более высокой частоты овариальных карцином.
При вторичном ДМ (ПМ) чаще наблюдаются неоплазмы органов половой сферы, легких, желудочно-кишечного тракта, почек. Реже встречаются лимфомы, саркомы, лейкоз, злокачественная меланома и т. д. Связь ДМ с опухолью четко аргументируется случаями излечения больных после удаления опухоли и, наоборот, рецидивом или прогрессированием заболевания при наличии метастазов.
Клинические проявления паранеопластического ДМ (ПМ) могут быть идентичны идиопатическому (первичному) ДМ, однако отмечается торпидность к проводимой терапии (стероидорезистентность), в том числе и к относительно большим дозам кортикостероидов.
В молодом возрасте сочетание ДМ (ПМ) с опухолью встречается редко, однако и в этих случаях необходима онкологическая настороженность, особенно при торпидном к лечению и атипично протекающем ДМ (ПМ).
Особое внимание следует обратить на возможность развития у этой группы пациентов лимфопролиферативных заболеваний, которые могут иметь свои особенности течения и маскировать более яркими критическими проявлениями ДМ (ПМ).
Таким образом, к «факторам риска» выявления злокачественных опухолей, помимо возраста и пола, следует отнести атипичное течение и рефрактерность ДМ (ПМ) к проводимой терапии, и наоборот – наличие миозитспецифических антител, которые практически не наблюдаются при опухолевом ДМ и ПМ. Нормальные значения креатинфосфокиназы (КФК) у больных с типичными проявлениями миозита могут также указывать на его связь со злокачественными новообразованиями.
Детский (ювенильный) дерматомиозит встречается приблизительно с одинаковой частотой у мальчиков и девочек, но, по данным некоторых авторов, может превалировать у мальчиков.
Е. Л. Насонов (2005) выделил ДМ (ПМ) у детей как особую форму заболевания в связи с выраженностью симптомов и частотой встречаемости васкулита. Соотношение ДМ и ПМ в этой группе примерно 2: 1. Выявляется бимодальное распределение ДМ/ПМ в детском возрасте: дети от 5 до 9 лет – 3,7 случая на 1 млн в год, детей от 10 до 14 лет – 4,3 случая на 1 млн в год. При начале заболевания до 7 лет отмечается его более легкое течение.
Прогноз ДМ в детском возрасте оценивается различно. Из 118 детей с ДМ (ПМ), наблюдавшихся Л. А. Исаевой и М. А. Жвания, 13 больных умерли, у 20 развилась тяжелая инвалидность, у остальных лечение сопровождалось улучшением состояния со снижением активности, а в ряде случаев и клинико-лабораторной ремиссией. У 10 больных длительность ремиссии составила 10 – 13 лет, что позволяет условно говорить о выздоровлении. В настоящее время с учетом более активной терапии, включающей пульс-терапию кортикостероидами, применение циклофосфамида, циклоспорина А и внутривенное введение иммуноглобулина (IgG), прогноз ювенильного ДМ (ПМ) значительно улучшился.
Клинические проявления ювенильного ДМ (ПМ) в целом сходны с таковыми у взрослых, однако имеются и некоторые особенности, связанные с выраженными васкулитами и микроангиопатиями, нередко более острым началом, экссудативным компонентом (отеки), синовитами с последующим развитием распространенного кальциноза тканей.
В большинстве случаев заболевание начинается с лихорадки, резкой боли в мышцах, кистях и стопах, нарастающей мышечной и общей слабости, прогрессирующего снижения массы тела.
Поражения кожи отмечаются у большинства больных и проявляются в виде лилового оттенка лица или характерной гелиотропной эритемы в периорбитальных областях, высыпаний в области лба, век, иногда щек, шеи, передней и задней поверхности грудной клетки, конечностей. Нередко одновременно развивается отек кожи, подкожной клетчатки, периартикулярных тканей, иногда имитирующий синовиты или действительно сочетающийся с ними. В области ногтевого ложа обнаруживаются микронекрозы (васкулит) и телеангиэктазии, а над суставами кисти – эритемы Готтрона с характерным цианотично-белесоватым оттенком, атрофией и восковидным шелушением. При тяжелых формах васкулита возможны изъязвления и некрозы кожи, висцеральных органов (кишечника и др.).
Поражение мышц характеризуется нарастанием мышечной слабости и обездвиженности больных, часто более выраженным болевым компонентом, что иногда трудно дифференцировать от полиартрита. Появляющиеся дисфагия и дисфония не позволяют сомневаться в диагнозе ДМ (ПМ). Особенно неблагоприятным фактором (симптомом) является нарастающее поражение дыхательной мускулатуры с развитием дыхательной недостаточности, аспирационной и гипостатической пневмонии. При более постепенном развитии клинической картины болезни вначале появляются небольшая мышечная слабость, иногда – кожные высыпания, умеренные синовиты и тендиниты, синдром Рейно. При поражении мышц тазового пояса ребенок часто падает, затем выявляется ограничение движений, развиваются стойкие контрактуры в суставах конечностей, атрофия мышц и, наконец, выраженная, иногда генерализованная кальцинация в области подкожной клетчатки и мышц. Все это приводит к почти полной обездвиженности и тяжелой инвалидизации больных ювенильным ДМ (ПМ). Кальциевые отложения появляются в среднем через 16 мес. после начала заболевания, изъязвляются, иногда инфицируются и при преимущественном отложении в области конечностей, тазового и плечевого пояса способствуют образованию контрактур, ограничению движений и иммобилизации больных в подростковом и молодом возрасте. Показано, что кальциноз развивается у 65 % не леченных стероидами пациентов. Механизмы кальцинации мало изучены, но их связь с предшествующим воспалением, васкулопатией и некротическими изменениями не вызывает сомнений. Обсуждается роль развития щелочной реакции и увеличения содержания щелочной фосфатазы в месте воспаления, локального повышения уровня кальция, фосфора и гликозаминогликанов. При этом лабораторные параметры фосфорно-кальциевого метаболизма остаются в норме.
Таким образом, ДМ (ПМ) у детей имеет определенные отличия:
1) наличие распространенного васкулита, проявляющегося клинически и особенно при морфологическом исследовании;
2) частое развитие подкожного кальциноза (в 5 раз чаще, чем у взрослых), что характеризует активный и прогрессирующий процесс развития заболевания;
3) редкое сочетание с опухолевым процессом.
Сочетание ДМ с другими заболеваниями соединительной ткани у детей наблюдаются сравнительно редко.
В. И. Мазуров и И. Б. Беляева (2005) выделяют следующие формы хронического течения ПМ: вариант Вагнера – Унферрихта (классическое течение), псевдомиопатическую, псевдомиастеническую, псевдоамиотрофическую, миосклеротическую, миалгическую и форму с синдромом Мак-Ардля.
Псевдомиопатическая форма ПМ развивается постепенно после переохлаждения, ОРВИ, ангины, иногда начинается с кратковременного повышения температуры тела до 37 – 38 °С. Формируются симметричные диффузные мышечные атрофии, возникают двигательные нарушения в проксимальных отделах конечностей, мышцах плечевого и тазового пояса с обязательным вовлечением мышц дистальных отделов верхних конечностей, рано развиваются фиброз и ретракции аддукторно-флексорно-ротатарной локализации. Болевой синдром не характерен. Мышцы лица не поражаются. Клинически наблюдается сходство с миопатией. При электромиографии (ЭМГ) выявляется миогенно-неврогенный тип поражения. Течение заболевания медленное, неуклонно прогрессирующее, приводящее к тяжелым двигательным нарушениям. Существует несколько вариантов его течения: по типу плечелопаточно-лицевой формы миодистрофии и конечностно-поясничных форм миопатий.
Псевдомиастеническая форма ПМ характеризуется сочетанием полимиозита с миастеническими явлениями. Нередко наблюдаются серьезные нарушения функций мышц лица, страдает мимическая мускулатура, поражаются дыхательные мышцы, развиваются дыхательные кризовые состояния вплоть до необходимости проведения трахеостомии. При ЭМГ выявляются неврогенно-миогенные признаки и утомляемость миастенического типа. Эта форма ПМ отличается тяжелым течением и плохим прогнозом.
Псевдоамиотрофическая форма ПМ характеризуется развитием диффузных мышечных атрофий, тяжелыми двигательными расстройствами, вплоть до невозможности передвигаться. У таких больных часто наблюдаются диспластические признаки – кифосколиоз, вогнутая форма грудной клетки. Отмечаются диффузная гипотония мышц и арефлексия. Иногда наблюдаются кожные высыпания по типу экссудативного диатеза, легкая пастозность лица. При ЭМГ выявляются миогенно-неврогенные признаки поражения. Течение тяжелое, иногда развивается полная обездвижимость.
Миосклеротическая форма ПМ встречается редко и характеризуется бурным развитием миосклероза и контрактур в начале заболевания. У пациентов формируются распространенные ретракции, фиксация конечностей, обездвиженность. В процесс вовлекаются дыхательные мышцы. Наблюдается поражение кожи. При ЭМГ выявляются миогенно-неврогенные изменения.
Миалгическая форма ПМ — встречается наиболее часто (34,9 % случаев). Чаще болеют женщины в возрасте от 15 до 45 лет. Основные проявления – сильные боли в мышцах и боли невралгического характера. Все симптомы ПМ, кроме болевого, представлены незначительно. Кожные покровы не поражаются. Страдает периферическая нервная система. Имеются эндокринные, вегетативные расстройства, арефлексия, полиневритический тип расстройств чувствительности. При ЭМГ выявляется преимущественно неврогенный тип изменений. Течение болезни благоприятное.
Форма с синдромом Мак-Ардля — редкий вариант полимиозита. Характеризуется развитием симптомокомплекса, имеющего сходство с гликогенозом V типа (болезнью Мак-Ардля), связанного с дефицитом мышечной фосфорилазы – фермента, участвующего в процессе гликогенолиза. При этом заболевании развивается ригидность в мышцах при физической нагрузке, сопровождающейся увеличением объема мышц, развитием утомляемости и слабости в сочетании с амиотрофическим синдромом, кожными проявлениями и признаками воспаления. При ЭМГ выявляется миогенно-неврогенный тип изменений.
Осложнения. Наиболее частое и грозное осложнение (занимает первое место среди причин смерти больных ДМ (ПМ)) – аспирация пищи при нарушении глотания и развитие тяжелой аспирационной пневмонии на фоне ограниченной подвижности грудной клетки вследствие поражения межреберных мышц и диафрагмы. Гиповентиляция легких также создает предпосылки к развитию пневмонии вследствие интеркуррентной инфекции. В отдельных случаях тяжелое поражение дыхательных мышц с резким ограничением экскурсии грудной клетки может вести к нарастающей дыхательной недостаточности и асфиксии, что требует применения ИВЛ. Сердечная и особенно почечная недостаточность при ДМ (ПМ) относительно редки. У обездвиженных больных часто возникают язвы, пролежни, которые легко инфицируются; возможны дистрофия, истощение.







